


Clinical Research Coordinator Certification In 2025

A Comprehensive Guide to Clinical Research Coordinator Classes
The medical field thrives on relentless innovation. Behind every groundbreaking treatment and medical solution is an intricate web of research, planning, and execution—and at the heart of it all is the Clinical Research Coordinator (CRC). If you've been considering a career in clinical research, becoming a CRC is an excellent way to be at the forefront of medical advancements.
Who is a Clinical Research Coordinator?
A Clinical Research Coordinator wears many hats. They’re the driving force behind research sites, ensuring that trials are executed seamlessly while maintaining regulatory and ethical standards. Often referred to simply as CRCs, these individuals manage documentation, safeguard patient well-being, and oversee the implementation of research procedures.
But this isn't a solo mission. CRCs operate under the guidance of a Principal Investigator (PI), the medical professional responsible for overseeing the clinical trial overall. Together, they collaborate to push scientific boundaries while maintaining strict compliance with research protocols.
For those ready to step into this crucial role, enrolling in Clinical Research Coordinator classes could be the first step towards launching your career.
What Does a Clinical Research Coordinator Do?
The day-to-day responsibilities of a CRC revolve around precision and professionalism. Whether managing trial logistics or ensuring participants’ safety, a CRC’s critical eye for detail is indispensable. Their duties often span months as clinical trials require careful planning, consistent monitoring, and extensive data collection.
Beyond technical skills, CRCs are also skilled communicators. They serve as representatives of their healthcare organization or institution, bridging gaps between research teams, sponsors, and participants. Building strong professional relationships is essential for the successful implementation of any study.
If you're organized, patient, and driven by the prospect of impacting healthcare innovation, the CRC role might just be your calling.
The Path to Becoming a Clinical Research Coordinator
There’s no single route to becoming a CRC, which means professionals from various educational backgrounds can find their way into this exciting field. Degrees and experience in areas like nursing, biology, pharmacy, business administration, statistics, medical technology, or teaching can all pave the way. CRCs are employed across a variety of settings, including research institutions, private companies, biotechnology firms, and pharmaceutical organizations.
Your Educational Foundation
While a bachelor's degree in microbiology, biology, or a related health science is often ideal, it’s not the only way to qualify for the role. Employers value relevant experience just as much as formal education. Completing coursework focused on clinical research will also give you a competitive edge when entering the industry.
Experience Matters
Many entry-level CRC positions require at least 1-2 years of professional experience in a healthcare or research environment. More senior roles demand anywhere from 5-6 years of experience and often a master's degree for leadership roles with greater responsibilities and higher pay.
The Role of Clinical Research Coordinator Classes
Formal education through Clinical Research Coordinator classes can lay the foundation for your future success. These programs are specifically designed to equip aspiring CRCs with practical knowledge, research expertise, and ethical training essential for excelling in the field. Here’s a breakdown of the types of CRC programs you can explore:
1. Certificate Programs
Focused and efficient, certificate programs offer a solid understanding of clinical research guidelines, ethics, and practices. These are often short-term and flexible, with some even offered online. Special courses, such as those in Pharmacovigilance or Good Clinical Practice (ICH-GCP), can enhance your qualifications further.
2. Associate’s Degree Programs
Looking for more comprehensive instruction? Associate’s degree programs take a deep dive into research methodologies, ethical considerations, and data management techniques. These take about two years and can include hands-on, practical training such as Clinical Trial Assistant programs.
3. Bachelor’s Degree Programs
A bachelor's degree in clinical research is the most in-depth option available. Spanning four years, these programs offer robust training in project management, regulatory compliance, and advanced research techniques. Graduates are well-prepared to step into senior CRC roles or even specialize further.
Why Take Clinical Research Coordinator Classes?
Enrolling in CRC classes isn’t just an investment in education—it’s an investment in your future. Here’s what you stand to gain:
Stronger Job Prospects
Employers favor candidates with formal education and demonstrated expertise. Classes set you apart as committed and prepared for the challenges of the role.Enhanced Skills and Knowledge
Gain proficiency in essential areas like regulatory compliance, clinical data management, and ethical research practices, all of which are crucial to a successful career.Opportunities for Career Growth
Advanced education often leads to better roles, higher salaries, and opportunities to contribute to groundbreaking medical trials.Networking Connections
Many programs connect you with industry professionals, instructors, and peers who can become valuable allies in your career.
For ambitious professionals, pursuing advanced certifications such as the Medical Monitor Certification or Advanced Principal Investigator Certification can lead to leadership roles in clinical research.
How to Choose the Right CRC Class
With so many options available, selecting the right program may feel overwhelming. Here are some factors to consider before making your choice:
Accreditation: Make sure the program is accredited by a credible organization to ensure quality education.
Curriculum: Review the course content to confirm it covers topics like research ethics, data analysis, and compliance with regulatory standards.
Learning Format: Decide whether online, in-person, or blended classes best suit your needs.
Cost and Time: Budget for tuition and assess how the program length fits within your timeline.
Final Thoughts
A career as a Clinical Research Coordinator offers a unique chance to contribute to medical breakthroughs that improve lives. With the right education, practical training, and dedication, you can make a meaningful impact in this dynamic field. Enrolling in clinical research coordinator classes will not only sharpen your skills but also open doors to new career opportunities.
Take charge of your future, invest in your education, and set your sights on a fulfilling career at the forefront of medical innovation.






Clinical Research Associate: A Full Guide on Becoming A CRA in 2025

Clinical Research Associate
A Complete Guide on How to Become a Clinical Research Associate
Whether you are new to this field or looking to advance your career, this article covers every aspect of becoming a successful CRA, including resources and practical advice for getting started.
What Does a Clinical Research Associate Do?
A Clinical Research Associate plays a crucial role in ensuring that clinical trials adhere to ethical standards, regulatory requirements, and Good Clinical Practice (GCP) guidelines. But don’t stop with the basics—understanding the nuances of the role will give you a professional edge.
Core Responsibilities of a CRA
Monitor Clinical Trial Sites
CRAs ensure trials follow protocols by conducting site visits and remote monitoring.Ensure Data Accuracy
They validate and review trial data to maintain precision and completion.Maintain Compliance
CRAs make sure that trials align with global regulatory guidelines, such as FDA, EMA, and ICH-GCP.Protect Participant Safety
Oversee participant accommodations, consent processes, and their overall well-being.Provide Professional Support
CRAs offer hands-on guidance and training to site staff to align their operations with trial protocols.
Lesser-Known Tip: Developing a working knowledge of decentralized trials and using digital platforms like wearable monitoring devices can set you apart as an innovative candidate.
What is CRA Meaning in Medical Context?
The CRA meaning medical focuses on professionals responsible for overseeing clinical research integrity and ensuring patient safety throughout the study process.
What Does a CRA Do?? They uphold the structure that makes groundbreaking medical advances possible.
Exploring the Best Clinical Research Associate Training Programs
Are you ready to jumpstart your career? The Best Clinical Research Associate Training Programs combine flexibility, practical knowledge, and real-world application.
Top Picks:
A fully customizable and self-paced course designed for both beginners and advanced learners.
IAOCR Certification Programs
Offers international focus, great for professionals interested in multi-national studies.
Ideal for aspiring CRAs who want to specialize in tech-forward monitoring.
Mayo Clinic IATA Training
Focuses on niche skills such as biological sample transport, making it a great complement to traditional CRA learning.
Lesser-Known Tip: Supplement your training with hands-on modules or internships for tangible accomplishments on your resume.
Understanding CRA Certification Requirements
Certifications are the hallmark of professionalism in clinical research. But what are the CRA certification requirements?
Here’s a breakdown:
Education Requirement
A Bachelor’s in life sciences is ideal (but not mandatory with prior experience or certifications).Training Completion
Enroll in programs like CRA Training Program to fulfill competency gaps.Monitor Hours
Certain certifications, like ACRAC Certification, may require logged hours of clinical trial monitoring.
Certifications like Clinical Research Associate Certification will elevate your role and value to potential employers.
How to Get Into Clinical Research without Experience
Breaking into clinical research can seem daunting, but the right approach can quickly get you noticed.
1. Certify First
Career shifts starting with practical certifications like the CCRPS Training Program, which provides you with the theoretical and practical foundations to excel.
2. Focus Your Search on Adjacent Roles
Investigate roles like research assistantships or clinical trial supporting positions as an accessible path to gain field experience.
3. Volunteer at Research Facilities
Clinical research hubs in academic programs often list facilities needing data entry or support contributions, bypassing conventional job postings.
Lesser-Known Tips for Aspiring CRAs
Expand Regulatory Knowledge Globally
Going beyond U.S.-focused compliance? Certifications such as IAOCR Certification cover global exposure.Master Technological Tools Early
Platforms for CRA Clinical Trials, like Medidata and Oracle CTMS, are becoming industry standards. Become fluent in these for an edge.Specialize in Niche Fields
Gene therapy, AI-related trial compliance, and wearable tracker technologies dominate emerging roles.
Pro Tip: Engage in targeted training through CRA Training Institutes for niche exploration options.
Primary Responsibilities of a CRA
Clinical Trial Monitoring: Overseeing the progress of trials to ensure they comply with protocols and regulatory requirements such as ICH-GCP.
Data Verification and Accuracy: Reviewing collected research data to ensure it aligns with trial objectives and has been recorded correctly.
Regulatory Audits: Preparing reports for regulatory agencies and ensuring site readiness for inspections.
Participant Safety Oversight: Safeguarding the rights, safety, and well-being of trial participants.
Example: A CRA monitoring a COVID-19 vaccine trial ensures all data related to vaccination doses and observed side effects are meticulously recorded.
A CRA wears many hats—site monitor, data validator, compliance officer—and their work directly influences the success of clinical trials. If you’ve asked yourself what is a clinical research associate or what does a CRA do, understanding these core responsibilities is key.
Is This Career Right for You?
Becoming a CRA might be the perfect role for individuals who:
Have a background in life sciences, nursing, or similar fields.
Possess strong organizational skills and attention to detail.
Are passionate about healthcare, technology, or pharmaceutical innovations.
Love a mix of office work and field visits, traveling to trial sites, and interacting with research professionals.
If you’re wondering about how to become a clinical research associate, we’ll cover that next.
How to Become a Clinical Research Associate (10 Step Guide)
Step 1. Obtain the Right Education
The foundation of your CRA career begins with education.
Suggested Fields of Study:
Biology
Pharmacology
Nursing
Health Sciences
A bachelor’s degree is typically required, but advanced degrees, such as a Master’s in Clinical Research, can set you apart from other candidates. For those from non-health-related fields, certifications like those offered by CCRPS can close the knowledge gap.
Hidden Tip: Highlight any coursework related to clinical research or data analysis on your resume to demonstrate your understanding of the industry.
Step 2. Understand the Role of a Clinical Research Associate
Before you take the next steps, it’s essential to understand the CRA's responsibilities.
What Does a CRA Do?
Coordinate Clinical Trials: CRAs monitor multiple sites to ensure trials comply with Good Clinical Practice (GCP).
Verify Data Accuracy: They ensure the integrity of collected trial data.
Ensure Compliance: CRAs monitor regulatory adherence from study initiation to completion.
Participant Safety: They guarantee participant safety and well-being throughout the trial.
This knowledge is critical for tailoring your resume and cover letter to align with recruiters’ expectations.
Step 3. Earn Specialized Certifications
Certifications like CCRPS Clinical Research Associate Certification are globally recognized and demonstrate credibility to potential employers.
Recommended Certifications:
CCRA Certification (ideal for those with previous monitoring experience).
Clinical Trial Associate Certification (a great stepping stone for entry-level candidates).
IAOCR Certification (helps in international trial compliance).
Certifications not only validate your qualifications but also make your application stand out.
Step 4. Enroll in a CRA Training Program
A Clinical Research Associate Training Program bridges the knowledge gap between academic learning and hands-on experience. These programs cover essential areas like GCP, site monitoring, and patient safety protocols.
Programs like CCRPS also offer flexible, self-paced modules perfect for those balancing other commitments.
Step 5. Gain Entry-Level Experience in Clinical Research
Start your clinical research career with roles like Clinical Research Coordinator (CRC) or Clinical Trial Assistant (CTA). These positions provide valuable on-the-job experience that translates directly to CRA responsibilities.
Quick Tip: During interviews, talk about your experience handling patient recruitment, managing regulatory documents, or coordinating trial procedures. These are key skills CRAs need.
Step 6. Network with Industry Professionals
Leverage conferences, social networks like LinkedIn, and training program alumni networks to make connections in the clinical research field. Training programs such as CCRPS often facilitate networking opportunities. These connections can open doors to mentorships and job interviews.
Step 7. Tailor Your Resume for a CRA Position
Your resume is often the first impression employers have of you. Highlight the education, certifications, and experience most relevant to the CRA role.
Key Components of a Strong CRA Resume:
Professional Summary: A concise pitch of your goals and qualifications.
Example: “Certified Clinical Research Associate with experience in site monitoring, compliance oversight, and data validation seeks to contribute to impactful healthcare advancements within a dynamic research organization.”Education and Certifications: List degrees and certifications such as CCRPS Certification prominently.
Experience: Focus on measurable accomplishments in clinical research roles or related positions.
Example: “Achieved 100% audit compliance across trial sites, ensuring seamless regulatory reviews.”Skills Section: Include keywords such as trial monitoring, GCP compliance, and risk-based monitoring.
Pro Tip: Use action verbs like “monitored,” “coordinated,” and “validated” for impact.
Step 8. Craft a Winning Cover Letter
Your cover letter should be tailored to highlight your unique skills and passion for the field of clinical research.
Cover Letter Example:
Dear [Hiring Manager’s Name],
I am writing to express my interest in the Clinical Research Associate position at [Company Name]. As a certified CRA with hands-on experience in trial site monitoring and regulatory compliance, I am eager to contribute to your organization’s mission of advancing medical research.
Throughout my career, I have excelled in coordinating multi-site clinical trials, ensuring adherence to GCP standards, and fostering collaboration between sponsors and investigators. My recent completion of the CCRPS CRA Certification has further honed my expertise in trial management and patient safety protocols.
I am enthusiastic about applying my skills to ensure seamless trial coordination at [Company Name]. Thank you for considering my application.
Sincerely,
[Your Full Name]
Quick Tip: Research the company’s recent trials or major projects and reference them in your letter to show genuine interest.
Step 9. Apply for CRA Jobs
Armed with your tailored resume and cover letter, start applying for CRA roles. Look for opportunities with Contract Research Organizations (CROs), pharmaceutical companies, or academic medical centers.
Where to Apply:
LinkedIn
Glassdoor
Pharmaceutical and CRO-specific job boards
Company career pages
Consider starting with internships or part-time roles if you’re transitioning into clinical research.
Step 10. Enhance Industry Knowledge
To thrive as a CRA, stay updated on industry trends and regulations. Certifications like the CCRPS CRA Training Program focus on niche areas like decentralized trials and wearable technology monitoring.
Proactively learning about evolving areas of clinical research can propel you to senior roles or specialized opportunities.
Step 11. Prepare for Interviews
Acing the CRA interview requires a mix of technical expertise and strong communication skills.
Common Interview Questions:
“How do you ensure compliance with GCP guidelines in your monitoring process?”
“What challenges have you experienced during audits, and how did you resolve them?”
“How do you balance maintaining safety and achieving trial goals?”
Use examples to demonstrate your problem-solving skills and ability to work under pressure.
Step 12. Plan Your Career Growth
The CRA role is a stepping stone to various advanced career paths.
Career Pathways:
Senior CRA: Lead complex, multi-site trials.
Clinical Project Manager: Manage trial strategies and oversee budgets.
Regulatory Affairs Specialist: Focus on compliance policies and regulatory documentation.
Stay ahead by completing advanced certifications or enrolling in specialized courses through CCRPS to continually evolve in your career.
Salary Overview for Clinical Research Associates
A major incentive for becoming a CRA is the attractive compensation package. The salary for Clinical Research Associates varies depending on experience, certification, and geography.
Entry-Level CRA Salary
Newly trained CRAs can earn:
United States: $60,000–$75,000 annually.
Europe: €45,000–€60,000.
Australia/Asia: Salaries average $45,000–$55,000 USD.
Experienced CRA Salary
With 3–5 years of experience, earning potential jumps to:
United States: $85,000–$120,000 annually.
Senior CRAs: Can command salaries of $130,000 and higher.
Adding Certifications Boosts Pay
Certifications like CCRA certification or completing training through CCRPS can significantly increase earning potential.
Did You Know? Some organizations pay even higher for CRAs specializing in niche fields like oncology or rare diseases.
When discussing the potential salary of a CRA, remember that variables such as educational background, additional certifications, and job location also play a role.
1. A Day in the Life of a Clinical Research Associate
Ever wondered what a typical day looks like for a CRA? While the work can vary depending on the trial phase and employer, here’s a snapshot of common activities:
Morning: Reviewing updates from clinical trial sites and preparing monitoring reports.
Midday: Visiting clinical research sites to audit records, ensure protocol compliance, and address any site issues.
Afternoon: Meeting with site staff to provide training or resolve trial-related concerns.
Evening: Logging findings and preparing documentation to send to the sponsor or regulatory authorities.
Example: A CRA monitoring a psychiatric drug trial may need to confirm patient consent forms are stored accurately and that participant follow-ups are conducted on schedule.
This blend of diverse tasks keeps the role engaging and varied.
2. Common Challenges Faced by CRAs and Solutions
While being a CRA is rewarding, the role is not without challenges. Here’s how to overcome some common obstacles:
Problem: Managing multiple sites simultaneously.
Solution: Use organizational tools like spreadsheets and calendars to track visit schedules and updates efficiently.Problem: Addressing site staff resistance to trial protocols.
Solution: Build rapport and provide ongoing training to site staff, emphasizing how compliance ensures trial success and participant safety.Problem: Staying updated on changing regulations.
Solution: Enroll in continuous education programs such as the CCRPS CRA Certification to stay informed.
By understanding potential challenges and preparing to address them, CRAs can excel in their roles.
3. Networking Tips for Aspiring CRAs
Building a strong professional network is essential for success as a CRA. Here’s how to make industry connections:
Leverage LinkedIn: Join groups for CRAs or clinical research professionals, and engage in discussions.
Attend Industry Events: Conferences like DIA Global or BIO International are excellent for meeting sponsors and CRO representatives.
Utilize Training Networks: Programs like CCRPS often provide networking opportunities with alumni and industry leaders.
Quick Tip: Prepare an elevator pitch about your skills and career goals when meeting potential mentors or employers.
Your network can open doors to job postings, mentorship, and valuable advice.
4. Technological Tools Used by CRAs
With clinical trials becoming more digitally driven, CRAs must be proficient in various tools:
Clinical Trial Management Systems (CTMS): Track trial status, site progress, and compliance.
Electronic Data Capture (EDC): Handle and verify patient data through platforms like Medrio or Oracle Clinical.
Remote Monitoring Tools: Analyze patient data in real-time through wearable technologies or remote systems.
Example: A CRA working on decentralized trials may use Real-Time Data Sharing (RTDS) platforms to monitor patient vitals and ensure study compliance remotely.
Programs like the CCRPS CRA Certification often include modules on mastering these tools, giving you a career advantage.
5. Pharma vs. CROs: Which Is Best for You?
Choosing between working for a pharmaceutical company or a Contract Research Organization (CRO) depends on your career goals:
Pharma Companies:
Typically offer higher salaries.
CRAs oversee fewer trials but may have longer timelines.
Focus on in-house training and development.
CROs:
Provide exposure to diverse therapeutic areas and trial phases.
CRAs work on multiple concurrent projects, building experience faster.
Often lead to better global networking opportunities.
If gaining varied experience is your priority, CROs might be more suitable. Alternatively, pharma companies are ideal for those who want long-term trial involvement.
6. Regulatory Bodies and Their Roles
Knowledge of regulatory bodies is essential for CRAs to ensure compliance. Here are key organizations to be familiar with:
FDA (U.S.): Monitors drug and medical device trials to ensure ethical standards.
EMA (Europe): European counterpart of the FDA, overseeing EU trials.
ICH: Offers harmonized guidelines for Good Clinical Practice (GCP).
WHO (Global): Issues ethical and operational guidelines for multinational trials.
Fun Fact: A CRA working in America would need to be well-versed in FDA protocols, while those in Europe focus on FDA and EMA differences.
Understanding these organizations enhances your ability to monitor trials effectively within varying jurisdictions.
7. Ethical Considerations in Clinical Trials
CRAs are responsible for upholding strict ethical standards in clinical studies:
Ensuring Patient Rights: Confirm that participants fully understand and consent to the trials they are part of.
Preventing Conflict of Interest: CRAs must ensure sponsors do not influence findings.
Adhering to GCP Principles: Ethical treatment of patients is non-negotiable.
A CRA’s commitment to ethics significantly impacts the credibility of a clinical trial’s outcomes.
Example: Suppose a CRA discovers a site is underreporting side effects. It’s their duty to investigate and intervene, prioritizing participant safety.
Programs like the CCRPS CRA Certification emphasize ethics through practical modules and case studies.
8. The Importance of Soft Skills
Being a CRA is not just about technical knowledge—it also requires essential soft skills:
Communication: Effectively convey trial updates to site staff and sponsors.
Time Management: Balance multiple responsibilities, including site visits, documentation, and reporting.
Problem-Solving: Quickly address disruptions or errors during trials.
Quick Tip: If you’re transitioning into clinical research and worried about how to become clinical research associate-ready, soft skills are as critical as certifications.
These traits enable CRAs to thrive in high-pressure environments.
9. Career Transitions and Growth
For many professionals, becoming a CRA is a stepping stone to advanced roles:
Regulatory Affairs Specialist: Contribute to drug approval processes.
Senior CRA or Lead CRA: Guide and train junior CRAs while overseeing larger projects.
Project Manager: Manage entire clinical studies, from planning to closeout.
CRPs are increasingly bridging to AI-driven analytics roles post-certification enhancing usability adaptive monitoring practices.
Programs like CCRPS provide pathways for advancing within the industry, making career growth seamless.
10. Global Opportunities for CRAs
Clinical trials occur worldwide, creating immense potential for CRAs to work in diverse locations and cultural contexts.
Global opportunities include:
Decentralized Trials: Monitor trials conducted across multiple countries remotely.
Specialized Roles: CRAs with niche skills, like oncology or pediatrics, are in demand globally.
Regulatory Liaisons: Help international studies comply with varying national guidelines.
By understanding global dynamics, you can position yourself as a highly versatile candidate ready for international projects.
Pro Tip: Combine certifications like CCRPS CRA Training with knowledge of regulatory standards like ICH-GCP to thrive in global roles.
Take charge of your career and watch your role transform into one of the most impactful positions in modern healthcare. Your future as a CRA starts now.
Top Training Programs for CRAs (Based on Reviews and Results)
Your success as a CRA depends on choosing the right clinical research associate training program.
1. CCRPS CRA Certification Training – Best Overall
Earned a reputation for its detailed modules, expert instruction, and hands-on approach. CCRPS allows students to:
Learn GCP principles, ethical considerations, and site monitoring techniques.
Access real-world case studies to prepare for industry challenges.
Benefit from job placement services to ease entry into the workforce.
2. IAOCR Certification Programs
Focused on international compliance, IAOCR is ideal for CRAs planning to work on multinational clinical trials.
3. IQVIA CRA Training
IQVIA stands out for integrating advanced data management and artificial intelligence into its curriculum.
4. Mayo Clinic IATA Training
This program emphasizes regulatory transport requirements for biological samples, catering to CRAs working in lab trials.
If you’re looking for the best CRA training programs, CCRPS consistently ranks at the top for flexibility and depth.
Frequently Asked Questions
What Qualifications Are Essential for Becoming a CRA?
Bachelor's degree in life sciences and certification from accredited programs like CCRPS.
How Long Does CRA Training Take?
Self-paced programs like CCRPS take 4–8 weeks, depending on the individual’s schedule.
What Is the Typical CRA Career Path?
Start as a Clinical Trial Assistant or Study Coordinator, gain certification, and transition to CRA roles.
Should I Get Certified?
Yes! Certifications like CCRA certification or a certificate from CCRPS make you stand out in job applications and provide essential skills for managing real-world trials.
Expert Tip: Include certifications in your LinkedIn profile to increase recruiter visibility.
What Is CRA Meaning in Medical Terminology?
CRA stands for Clinical Research Associate, a critical role in clinical trials ensuring compliance with protocols and regulations.
For anyone asking what is CRA certification and its benefits, certifications validate your ability to oversee scientific studies ethically and efficiently.
Advanced Opportunities for CRAs
CRAs have unique opportunities for growth and specialization, with career advancement options including:
CRA Team Manager: Overseeing a team of CRAs across multiple sites.
Clinical Project Manager: Managing entire clinical studies from planning to completion.
Regulatory Affairs Specialist: Moving into regulatory oversight roles for new trials.
CRAs specializing in newer fields like gene therapy or AI-integrated clinical trials are in especially high demand.
Example: A Senior CRA role for a multinational oncology trial can earn you $150K+ per year.
Why Choose CCRPS for CRA Training?
The CCRPS CRA Certification Program delivers unmatched value:
Comprehensive training modules tailored to current industry needs.
Flexible self-paced learning for convenience.
Career advancement tools and networking opportunities to break into the industry.
Enrolling with CCRPS provides everything you need to become a confident, skilled CRA ready to tackle the challenges of 2025.
The pathway to becoming a Clinical Research Associate in 2025 is filled with opportunities to grow professionally and contribute meaningfully to global healthcare. With the right training, certifications, and experience, you could be monitoring innovative clinical trials that shape the future of medicine.
Take the first step by enrolling in the CCRPS CRA Training Program today—prepare for a rewarding and impactful career in clinical research.
Starting a career as a Clinical Research Associate (CRA) opens doors to medical innovation, competitive pay, and meaningful opportunities. With over 1.9 million science graduates each year, many face hurdles like entry-level roles requiring 1–2 years of experience or demanding lab work that falls short of career expectations. Clinical research training offers a fast-track solution.
Why Become a CRA?
CRAs ensure clinical trials are conducted ethically and efficiently, working alongside physicians and medical teams. Whether you’re aiming for experience before medical school or a new path entirely, CRA roles combine management and medical research into a dynamic, fulfilling career.
Key Benefits of a CRA Career
High Earnings: Certified CRAs earn $4,500–$12,000/month, with a 33% annual promotion rate.
Flexibility: Remote roles and opportunities to travel—expenses covered.
Career Growth: Start as a Clinical Trial Assistant or Coordinator and quickly advance with certification.
Get Ahead with CCRPS Training
CCRPS has 8 years of proven graduate success and resources institutions alone cannot provide due to our career and advanced education focus.
Fast-Track Timeline: Earn certification in as little as 4 weeks.
No Experience Needed: Our course prepares you for entry-level roles with no prior clinical experience required.
Expert Guidance: Learn from experienced professionals while building key industry connections.
Clinical Research Associate Certification: Your Path to a Rewarding Career
Are you a foreign doctor ready to make an impact in the U.S. healthcare system or seeking a high-growth, science-focused career? Becoming a Clinical Research Associate (CRA) could be your perfect solution.
Why Pursue a CRA Career?
Leverage Your Medical Expertise
Foreign doctors with medical degrees, such as MBBS, can bypass the challenges of USMLE and residency by transitioning into CRA or Clinical Research Coordinator (CRC) roles. Put your medical knowledge to work in a fulfilling alternative career.Gain a Distinct Skillset
CRA training provides expertise in conducting and managing clinical trials, adhering to ethical guidelines, and ensuring Good Clinical Practice (GCP). For a strong foundation, explore the ICH-GCP Course to master industry essentials.
The Most Comprehensive CRA Training with 196+ Modules
Our course is now even more robust, featuring over 196 modules—making it the most comprehensive CRA certification program available online. Designed by industry veterans, this curriculum ensures you're not just job-ready but set up for long-term success.
Path to Strategic Career Growth
General courses often fall short, leaving graduates unprepared. Our advanced training covers Senior CRA-level expertise, enabling you to secure coveted positions in top pharmaceutical companies, academic institutions, or research organizations. Interested in additional roles? Check out programs like the Clinical Trials Assistant Training for entry-level pathways or the Advanced Principal Investigator Certification for leadership opportunities.
Diverse Opportunities in a Thriving Industry
The clinical research field offers wide-ranging and flexible career options, from working with pharmaceutical giants like Pfizer to managing trials at prestigious academic institutions. Expand your knowledge in specialized areas through Pharmacovigilance Certification or Medical Monitoring Training to further enhance your career.

Clinical Research Associate Certification Qualifications
Foreign Doctors Welcome: A Clinical Research Associate or Coordinator plays a vital role in directing and supervising clinical trials conducted by physicians, nurses, and other science professionals. This career path is particularly attractive to many foreign doctors with completed medical degrees (MBBS) who can utilize their expertise in the US healthcare system by pursuing a CRA career instead of taking the USMLE or repeating residency training. For those interested in coordinating aspects, consider the Clinical Research Coordinator course.
Distinct Skillset: Unlike the traditional medical field you may be familiar with after years of schooling, Clinical Research Associate training provides a distinct and valuable skillset. For comprehensive understanding of Good Clinical Practice, see the ICH-GCP course.
Most Extensive Online Course: Our program goes beyond basic introductions, offering a comprehensive curriculum with over 110 modules – the most extensive Clinical Research Associate course available online. This in-depth training ensures you're well-prepared to secure a coveted CRA position.
Superior Coursework: Securing a CRA role is a strategic career move compared to the limitations of many traditional medical positions. While generic courses abound, we've observed that graduates often struggle due to a lack of substantive content. Our Clinical Research Associate course addresses this gap by providing Senior Clinical Research Associate-level training through 110 intensive modules grounded in the latest scientific principles. For those looking to assist in clinical trials, the Clinical Trials Assistant Training may also be of interest.
Diverse Career Opportunities: This high-demand science-based medical field offers diverse opportunities:
Work in the Private Sector: Pursue a CRA career with renowned pharmaceutical companies like Pfizer. Enhance your skills with the Advanced Clinical Research Project Manager Certification.
Academic Opportunities: Work in the academic sphere at medical schools. Those aiming for higher responsibilities may consider the Advanced Principal Investigator Physician Certification.
Unmatched Flexibility and Knowledge: In addition to our exceptional course content, we boast the largest number of clinical research courses available online, providing you with unmatched flexibility and knowledge. For those interested in safety monitoring of drugs, the Pharmacovigilance Certification and Medical Monitor Certification can enhance your capabilities in these critical areas.
Why Take A CRA Certification Course
Hidden Opportunities with Clinical Research Associate (CRA) Certification
A Clinical Research Associate (CRA) certification isn’t just about qualifying for a job—it’s about positioning yourself in one of the most dynamic and enriching career fields in healthcare. If you’re intrigued by clinical trials and medical innovation, here are some lesser-known but invaluable insights into the CRA certification process and its impact.
What Makes CRA Certification Unique?
Eligibility for Global Job Markets
A CRA certification opens doors not only in the U.S. but also in global markets. Clinical trials are conducted worldwide, and many pharmaceutical giants outsource research to international locations. Being certified with globally recognized credentials (like ACCRE) makes you competitive for roles in Europe, Asia, and beyond.Direct Contribution to Drug Safety
Did you know a CRA is instrumental in uncovering rare side effects of new treatments during trials? Their role in adverse event reporting ensures the safety of future drugs and devices. This critical function showcases how a CRA impacts healthcare on a global scale.Regulatory Expertise Across Borders
Certification programs don’t just teach you about Good Clinical Practice (GCP) guidelines—they prepare you to adapt to various regulatory frameworks like ICMRA standards or EU Clinical Trial Regulations, which vary from country to country.
Unrivaled Flexibility in Learning
Integrated Microlearning Modules
Certified programs now include bite-sized, module-based learning that accommodates even the busiest professionals. You can tackle complex topics in short, manageable segments, making learning less overwhelming without sacrificing depth.Simulated Trial Experiences
Some advanced CRA courses offer virtual clinical trial simulations. This allows you to experience trial management and monitoring in real-world scenarios, preparing you to hit the ground running.Personalized Career Development
Top-tier courses now include career mentorship programs, guiding students through networking, resume building, and interview strategies.
Jumpstart a Career Without Traditional Barriers
No U.S. Healthcare Background? No Problem
Many foreign professionals assume clinical research jobs demand U.S.-based experience. However, CRA certification provides all the foundational knowledge required to transition into the field and bypass requirements like USMLE or additional residency.Open to Non-Medical Professionals
While a healthcare or life sciences degree is beneficial, CRA certification is increasingly accessible for individuals with adjacent professional experience, like in project management or quality assurance. Some programs provide bridge courses for those without clinical backgrounds.Accelerate Your Career with Soft Skills
Beyond hard skills, certification ensures you develop essential soft skills like communication, data management, and attention to detail—critical factors often overlooked by non-certified applicants.
Lucrative and Less Explored Career Paths
Pharmaceutical Partnerships
CRAs can work directly with sponsors like Novartis or Amgen, managing trials that test cutting-edge therapies such as gene-editing drugs. These projects offer a front-row seat to medical breakthroughs.Remote CRA Roles
Few know that many CRA positions are 100% remote, cutting down the need for constant travel. Virtual monitoring of trials has become standard in many companies thanks to advanced technology.Niche Opportunities in Rare Disease Trials
Specializing in rare disease clinical trials connects you with groundbreaking projects where CRAs play a larger role due to the smaller teams involved. This niche often provides higher earning potential.
Behind-the-Scenes Secrets of CRA Certification
Specialized Certification Bonus
Many companies offer salary bonuses for obtaining specialized certifications alongside CRA credentials, such as Pharmacovigilance or Medical Monitoring.Direct Path to Leadership
Certification isn’t the endpoint; many certified CRAs transition into advanced roles like Clinical Research Managers or Project Leaders within three to five years.Boost Your Job Prospects
Even with general certification, including training from an ACCRE-accredited program, you'll join a network of former students, providing insights into hidden job openings and employer preferences.
Advanced Certification for Diverse Opportunities
Pairing CRA certification with additional qualifications expands your versatility:
Clinical Trial Auditing Certification lets you oversee trial compliance for top regulatory agencies like the FDA or EMA.
Oncology-Specific Training equips you to manage trials in one of the fastest-growing fields in medicine.
The Best CRA Certification Course for Entry-Level Professionals
Breaking into the field of clinical research as a Clinical Research Associate (CRA) can feel daunting, especially at the entry level. There’s a significant demand for well-trained CRAs, but companies are often hesitant to hire individuals without a strong background or certification. Why? The stakes are high—patient safety and trial integrity depend on skilled monitors. That’s where proper training becomes essential.
If you’re searching for a rigorous, results-driven CRA certification course that equips you with all the essential skills to excel, here’s everything you need to know.
Why Choose the CCRPS CRA Certification Course?
The CCRPS CRA Certification Course has been consistently recognized as one of the most comprehensive and effective clinical research programs online. Tailored for entry-level candidates, it bridges the experience gap with in-depth, hands-on learning that prepares students to hit the ground running.
What Sets This Course Apart?
Comprehensive Curriculum
Unlike many courses offering only 5-20 interactive modules, CCRPS provides over 196 densely packed modules covering every facet of clinical research monitoring. Topics include regulatory guidelines, protocol adherence, monitoring techniques, ethical considerations, and real-world scenarios you may encounter on the job.Advanced Training Beyond Basics
While other platforms focus on cursory introductions, CCRPS trains all students at a Senior CRA level, regardless of their prior experience. The material is designed to not only help you ace job interviews but also to excel in your role once hired.Expert-Led Instruction
The program is taught by seasoned industry professionals with over 15 years of hands-on CRA experience. They bring invaluable insights from the field, and students can ask questions privately after each session, ensuring personalized guidance.Guaranteed Preparedness
From learning how to interpret complex medical protocols to mastering compliance with regulatory standards, CCRPS equips you with the skills you need to succeed. Students are also trained to handle challenges specific to clinical research, such as managing adverse events and ensuring trial integrity.Success Rate You Can Trust
Currently, an impressive 82% of CCRPS graduates secure CRA positions within the first month of completing the course.Optional Internship Experience
For candidates with limited experience or those looking to strengthen their resumes, CCRPS offers a 6-month remote internship opportunity. This hands-on training ensures that you gain real-world exposure while actively applying for jobs.Flexible Formats
The program is fully online, making it accessible for professionals with busy schedules. Despite its remote format, the course fosters interaction through discussion forums, mentorship, and a curriculum designed for immersive online learning.
Who Can Take the Course?
The course welcomes a diverse range of professionals, including science graduates, nurses, and even physicians looking to transition to clinical research. No prior CRA experience? No problem. The course is specifically designed to train and certify individuals from various backgrounds, ensuring that everyone receives the same advanced-level education.
Unique Insights Into CRA Certification
Medical Knowledge Isn’t Enough
Although medical professionals often transition into CRA roles, medical knowledge alone won’t suffice. Clinical research monitoring requires a deep understanding of protocols, guidelines, and ethical considerations, which this training focuses on.Standout Feature
The program includes all potential scenarios you might face as a CRA, arming you with the tools to handle even the most complex trials.Industry-Recognized Accreditation
CCRPS is ACCRE-accredited, a hallmark of quality and industry recognition.
Career Advice and Job Placement Resources
After certification, the next step is securing a position. While CCRPS does not directly place students in jobs, it offers invaluable guidance and resources to help you succeed in the job market.
Professional Resume & Cover Letter Support
TopCV: Upload your resume for a free review and feedback that improves your chances of standing out.
ResumeRabbit: Distribute your resume to over 60 job boards, saving you time and increasing visibility.
Recruiting & Job Boards
Clinical Research Guidance
Clinical Trial Podcast with Kunal: Provides insights into the clinical research industry and advice for navigating your career.
Networking Opportunities
Engage with online CRA communities or LinkedIn groups dedicated to clinical research. Many jobs aren’t advertised publicly, so networking can open doors to hidden opportunities.
Application Tips for CRA Jobs
Tailor Your Applications
Always customize your CV and cover letter for each specific role and company. Use the job description as a guide to highlight relevant skills and experiences.Follow Up
If you don’t hear back, don’t assume the application was rejected—it may have been overlooked. Follow up to ensure your application gets the attention it deserves.Showcase Your Certification
Mention your CCRPS certification prominently, detailing key skills you gained from the course that align with the job requirements.Highlight Internship Experience
If you’ve taken advantage of CCRPS’s internship, list your responsibilities and achievements to demonstrate hands-on experience.
Resume Writing Services
ResumeGo - Offers industry-specific, keyword-optimized resumes with one-on-one consultations and unlimited revisions. Learn more.
Let’s Eat, Grandma - Provides tailored resume writing services with a focus on detailed consultations. Explore options.
TopResume - Known for its free resume review and emphasis on keyword optimization. Check it out.
Job Boards for Clinical Research
PharmiWeb.Jobs - A comprehensive job board for clinical research roles, including Clinical Research Associate positions. Visit PharmiWeb.
HCSRN Job Board - Lists faculty and staff positions at research centers and academic institutions. Explore HCSRN.
Networking Opportunities
LinkedIn Groups - Join groups like "Clinical Research Professionals" to connect with peers and discover job opportunities.
Clinical Trial Podcast - Offers insights and advice for navigating the clinical research industry. Listen here.
Exploring the ICH-GCP in Clinical Research
Clinical research is more than just testing new treatments—it's about ensuring the safety and dignity of participants while generating data that is trustworthy and accurate. To achieve this, clinical studies globally adhere to the ICH-GCP guidelines—the International Committee for Harmonization of Good Clinical Practice. These guidelines set the gold standard for safeguarding human participants' rights and integrity while maintaining the validity of the research process.
Why are these standards so important? For one, they ensure clinical trials respect the rights and dignity of every patient involved. Secondly, they outline how data should be collected, securely stored, and analyzed to ensure accuracy. Regulatory bodies, such as the FDA in the U.S., have enforced compliance with these guidelines, including detailed protocols like the FDA’s E6(R2) Guidance for Good Clinical Practice.
Sticking to these international standards isn’t just good practice—it’s essential for anyone pursuing a career in clinical research. If you’re aspiring to make your mark as a Clinical Research Associate (CRA), understanding the ICH-GCP is your starting point.
What Does It Take to Become a CRA?
A CRA’s role involves monitoring clinical trials, ensuring proper protocol adherence, and safeguarding participant safety. But what skills and qualifications do you need to thrive in this career path?
Educational Foundation
A degree in a biological science, medicine, nursing, or life sciences is commonly required. Degrees in fields like pharmacology, biochemistry, or immunology are particularly valuable.Technical Expertise
CRAs are expected to manage essential data systems like Electronic Data Capture (EDC), handle regulatory documents, and analyze clinical trial data.People Skills
Equally crucial is emotional intelligence. Strong interpersonal skills are non-negotiable since CRAs often coordinate between various teams, troubleshoot issues, and guide study sites towards compliance.
Beyond technical and interpersonal expertise, being detail-oriented and possessing a thorough understanding of medical trial regulations are vital traits. The ability to spot compliance violations and implement corrective actions ensures the success and integrity of the study.
Building Your Career Path as a CRA
Gone are the days when medical or nursing professionals could transition into CRA roles without research training. Today, most employers seek individuals with direct clinical research experience or specialized training. While Clinical Trial Assistants (CTAs) and Clinical Research Coordinators (CRCs) often receive preference, aspiring CRAs now have access to professional courses that open career doors.
For instance, master's programs in clinical research offered by universities are a solid option but can take years to complete. Those who want a faster yet comprehensive route can explore certification programs designed specifically for CRAs—programs like the Advanced Clinical Research Associate Certification (ACRAC).
A Toe in the Door for Non-Clinical Professionals
Think you can’t become a CRA without prior experience or a specialized degree? Think again.
The CCRPS ACRAC program is one of the most accessible courses for candidates without a clinical research background. What makes it unique?
Comprehensive Curriculum
With over 196 high-impact modules, the program dives deep into every possible scenario you could face as a CRA. The training equips you to master regulatory requirements, research ethics, trial protocols, and compliance standards.Accessible to Fresh Graduates
You don’t need years of lab work or experience as a Clinical Research Coordinator. With just a B.S. degree in a life science, this program prepares you to step directly into a CRA role.Hands-On Guidance
Taught by experts with 15+ years in the field, the program offers personalized support and mentorship, helping you polish your skills and readiness for job interviews.Industry Accreditation
The ACRAC program carries multi-accreditation from respected institutions like ACCRE, ACCME, and ANCC, giving your certification global recognition.Optional Remote Internship
To boost your resume, CCRPS even offers an internship option, allowing you to gain hands-on experience while applying for jobs.
These opportunities make the ACRAC program an unparalleled option for breaking into clinical research.
Your Next Step
A career as a Clinical Research Associate offers not only competitive salaries but a chance to directly contribute to life-saving medical innovations. With the growing demand for CRAs and the right training, you can kickstart your career without waiting for years of experience.
Take the leap today. Enroll in the Advanced Clinical Research Associate Certification (ACRAC) and ensure your place in this exciting, impactful field.
Elevate Your Career in Clinical Research with CCRPS ACRAC
If you’ve been dreaming of becoming a Clinical Research Associate (CRA) or want to take your clinical research career to the next level, the CCRPS Advanced Clinical Research Associate Certification (ACRAC) is the definitive program for comprehensive, hands-on training. This meticulously structured program doesn’t just set the gold standard—it ensures you’re fully prepared for one of the most rewarding, impactful careers in healthcare and research.
Here’s an in-depth look at how the ACRAC program sets you up for success.
Flexible and Achievable Learning
Studying for a new career shouldn’t mean putting your current life on hold. With the ACRAC program’s self-paced structure, you have the freedom to choose how and when you study. The program offers approximately 250 hours of in-depth training content, spread across 196 modules that build your competence step-by-step.
For students dedicating full-time focus to their studies, certification can be achieved in just two to three weeks. But for those balancing other responsibilities, you can tailor the program to your rhythm and study at your own pace. This flexibility ensures access to a robust educational foundation without unnecessary pressure.
An Exhaustive Curriculum Covering Every CRA Competency
The breadth and depth of the curriculum are unmatched. With 196 detailed modules, the ACRAC program ensures you acquire expertise across every key area of clinical trials and research monitoring. Here’s how the training is structured:
Core Foundations
Introduction to clinical research terminology, concepts, and abbreviations.
Overview of the principles of Good Clinical Practice (GCP), informed by both ICH and FDA guidelines.
Understanding the regulatory framework and compliance obligations, including FDA E6(R2) guidelines and key CFR sections.
Ethics and Compliance
Comprehensive training in designing trials that meet ethical standards for human subjects, particularly for vulnerable populations such as children, pregnant women, and individuals with impaired consent capacity.
Understanding informed consent procedures, minimizing coercion, and maintaining transparency with trial participants.
Clinical Trial Protocol Development
Step-by-step guidance on the creation, implementation, and documentation of Clinical Trial Protocols (CTPs) in compliance with ICH-GCP standards and federal regulations.
Training in logistical essentials, such as IRB/IEC approval processes and navigating trial budgeting and funding.
Pharmacovigilance and Adverse Event Management
Instruction on identifying, documenting, and managing Adverse Events (AEs), with distinctions between different categories like Adverse Drug Reactions (ADRs) and Serious Adverse Events (SAEs).
Mastering causality assessments for adverse events and ensuring clear communication with IRBs and sponsors.
Site Visits and Trial Monitoring
Thorough preparation for all site visit types, including:
Site Qualification Visits (SQVs) for assessing site readiness and investigator qualifications.
Site Initiation Visits (SIVs) for introducing site staff to trial protocols and regulatory expectations.
Routine Monitoring Visits (RMVs) for supervising ongoing trial compliance.
Close-Out Visits (COVs) to ensure all documentation and compliance tasks are complete post-trial.
Extensive focus on documentation, including Case Report Forms (CRFs), Trial Master Files (TMFs), and Source Data Verification (SDV).
Tools and Technology
Mastery of digital tools used in modern clinical trials, such as Electronic Data Capture (EDC) systems, Interactive Response Technologies (IRT), and remote monitoring software.
Training in adopting new and evolving technologies, including risk-based monitoring (RBM) and centralized monitoring practices.
With each module, you’ll build a clear understanding of both the theoretical foundations and the practical application of clinical trial protocols. The program’s thoughtful design ensures emerging CRAs are equipped to seamlessly integrate into the workforce.
Real-Life Problem Solving for CRAs
The ACRAC program goes beyond theoretical training. Through realistic scenarios and case studies, you’ll learn how to handle critical challenges that CRAs face on the job, such as:
Identifying protocol deviations and violations, escalating when necessary, and working with study teams to implement corrective actions.
Dealing with ethical misconduct or research fraud without compromising the integrity of trials.
Maintaining the delicate balance of safeguarding participants’ rights and ensuring smooth trial progress.
These hands-on exercises ensure you’re ready to tackle unforeseen challenges, making you a valuable asset to any clinical research team.
CME Credits for Career Growth
The ACRAC program doesn’t just certify you—it enhances your professional credentials. It provides 17.5 Continuing Medical Education (CME) credits, recognized by prestigious institutions like the Accreditation Council for Clinical Research & Education (ACCRE). These credits can boost your resume and help you build expertise in other healthcare-related industries like medicine, nursing, or pharmacy.
Why ACRAC is Ideal for Aspiring CRAs
Whether you’re fresh to the field or pivoting to clinical research from another career, ACRAC is tailored to meet your unique needs. Unlike many programs, it assumes no prior experience in clinical research. Instead, it focuses on teaching you everything you need to know to perform at the level of a senior CRA.
With certifications from globally respected bodies like ACCME and Transcelerate Biopharma, the program ensures your qualifications are recognized internationally, opening a world of job opportunities.
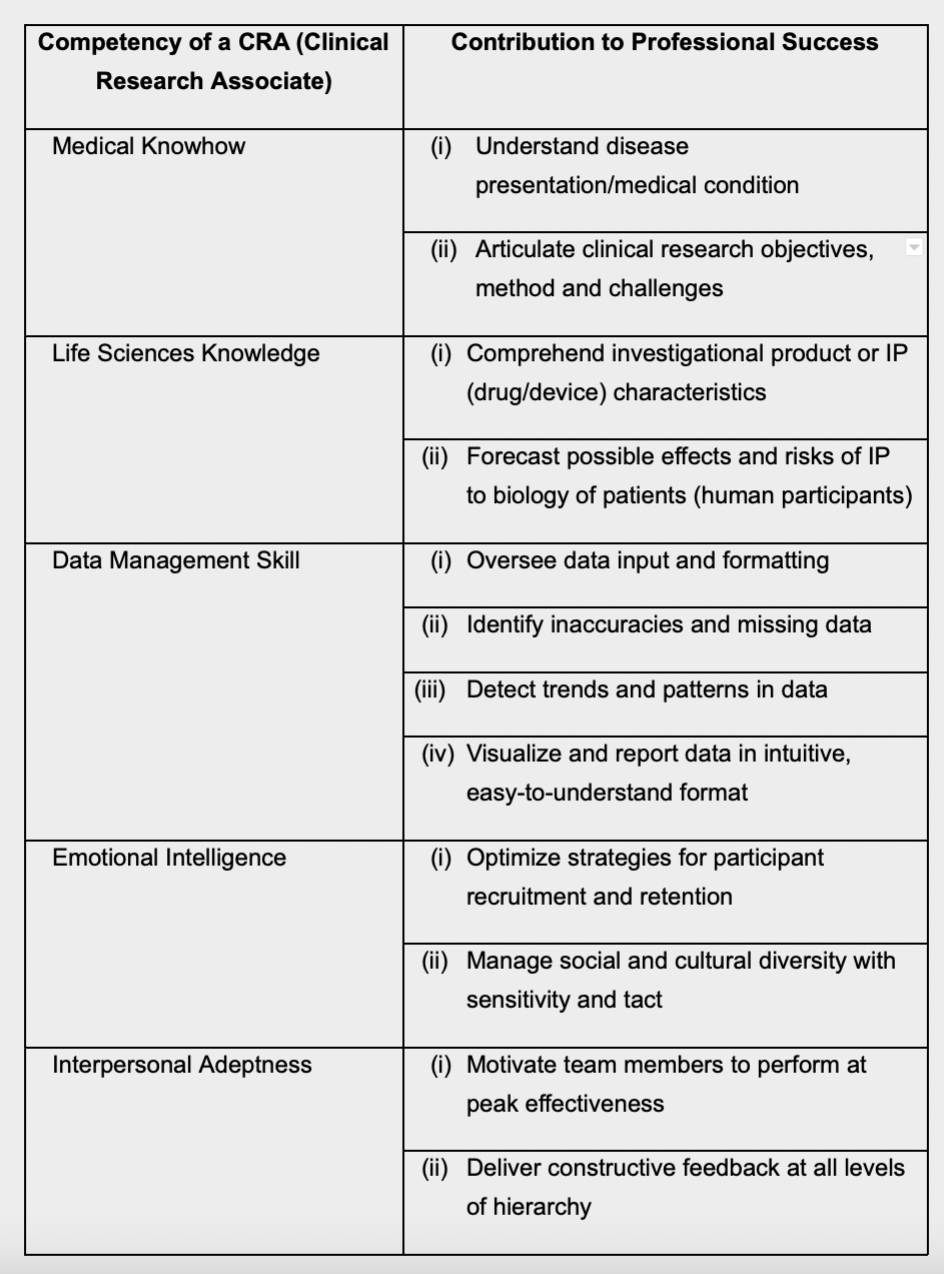
Core Competency Framework for CRAs
To illustrate, the ACRP’s ‘Core Competency
Framework for Clinical Study Monitoring’
requires that a CRA should be able to identify
and correct compliance violations at a study
site. The CRA must not only bring such
violations to the attention of site staff, s/he
must induce them to take corrective action,
as well as reporting the matter and even
escalating it, where necessary14.
The table below summarizes the ideal
competencies of a CRA, and provides
insights on how each ability contributes to
the CRA’s performance.

A Comprehensive Look at the ACRAC Syllabus for Clinical Research Associate Training
The CCRPS Advanced Clinical Research Associate Certification (ACRAC) syllabus is an exceptional, all-encompassing program meticulously crafted to provide aspiring Clinical Research Associates (CRAs) with the knowledge, expertise, and tools to thrive in the clinical research industry. It’s an extensive syllabus, not just a training program—it’s a launching pad for an impactful career in clinical research.
With 196 thoughtfully curated modules, the ACRAC program dives deep into every competency and skill required for CRAs at every stage of their career. Whether you’re new to the field or looking to enhance your expertise, this syllabus ensures you’re fully prepared to monitor and manage clinical trials with confidence and professionalism.
Foundations to Advanced Clinical Monitoring
The program kicks off with a solid foundation in clinical research essentials. Through the Introduction section, you’ll get acquainted with the critical roles and responsibilities of a CRA, the principles of Good Clinical Practice (GCP), and how ethical and legal standards govern the clinical trial landscape.
Stakeholder Collaboration: You’ll explore how CRAs collaborate with sponsors, IRBs, investigators, and site staff to ensure harmony between regulatory requirements and trial objectives.
ICH-GCP Overview: This part unpacks global guidelines, like the ICH GCP and FDA’s standards, crucial for the smooth execution of clinical trials, including a forward-looking discussion on trends in monitoring.
Sponsor and Investigator Roles
Moving into the Sponsor and Investigator Roles modules, the syllabus deepens your understanding of the complex partnerships in clinical research. Covering key sections of ICH GCP (E6), you’ll learn about reporting responsibilities and the critical measures sponsors and investigators must take to maintain participant safety, data validity, and compliance.
These modules are particularly valuable for CRAs tasked with maintaining open communication between parties and ensuring no stone is left unturned in fulfilling regulatory expectations.
Clinical Trial Phases and Procedural Expertise
The program exhaustively reviews all phases of clinical trials—from preclinical to post-marketing studies—with a focus on monitoring techniques tailored to each phase’s unique demands. Highlights include:
Safety Compliance: Learn best practices for ensuring trial safety and managing unexpected challenges during monitoring visits.
Trial Design Techniques: Mastering protocol-specific methodologies to ensure data accuracy and participant welfare.
Specialized Trials: You’ll gain insights into handling trials involving vulnerable populations, including children and individuals with cognitive disabilities. These modules prepare you to manage ethically sensitive scenarios with confidence.
Advanced Clinical Trial Protocol Development
The importance of a well-crafted Clinical Trial Protocol (CTP) cannot be overstated. The syllabus dedicates an entire section to guiding you through the drafting, reviewing, and assessing process. You’ll learn how to define inclusion and exclusion criteria while addressing common complexities, such as trials involving older adults or pregnant women.
Protocol Deviations and Violations: Handling major and minor deviations is a skill every CRA needs. You’ll learn how to identify, mitigate, and resolve these issues effectively.
Site Monitoring Visits
CRAs are the backbone of site monitoring, a responsibility that requires impeccable preparedness and communication. The syllabus provides unparalleled training on all types of monitoring visits:
Site Qualification Visits (SQVs): Get step-by-step guidance for pre-study checks, investigator selection, and evaluation.
Site Initiation Visits (SIVs): Learn how to prepare documentation, educate site staff, and communicate protocols to ensure a strong trial launch.
Routine Monitoring Visits (RMVs): Gain expertise in ensuring compliance, tracking progress, and correcting deficiencies during active trials.
Site Close-Out Visits (COVs): Equip yourself with tools to confirm proper data submission, secure records, and leave no loose ends upon trial completion.
Practical templates and checklists accompany these modules, so you hit the ground running from day one.
Modernized Monitoring Strategies
Keeping up with advancements in technology is a major component of the ACRAC program. Tackling everything from electronic data capture (EDC) systems to risk-based monitoring (RBM) approaches, you’ll learn how modern tools enhance trial efficiency and data accuracy.
Specialized modules on remote and centralized monitoring, which became especially critical during the COVID-19 shift, ensure you’re up to date with the latest methods.
Pharmacovigilance and Regulatory Affairs
Safety is at the heart of clinical research, and the program’s focus on pharmacovigilance ensures you’re prepared to monitor adverse events (AEs) and maintain compliance with regulatory requirements.
Adverse Events Reporting: From causality analysis to periodic updates, you’ll master the protocols for identifying, documenting, and reporting safety concerns.
Investigational Products: Learn storage, inventory, and accountability best practices for medical products in a way that ensures full traceability.
Specialized Monitoring Techniques
The syllabus dives into niche areas of monitoring, including oncology, gene therapy, and rare disease trials. These specialized modules will prepare you to adapt to evolving demands of unique trial types and patient populations.
Leadership, Team Management, and Advanced Strategies
Leadership and decision-making skills are integral to a CRA's role. The syllabus equips you with strategies to manage cross-functional teams, negotiate with sponsors, and build collaborative relationships with stakeholders.
The advanced strategy modules elevate your skills further, offering insights into modern methodologies like adaptive monitoring, predictive analytics, and machine learning applications in trial oversight. These cutting-edge tools are essential for data-rich environments in global trials.
Patient Recruitment, Retention, and Misconduct Prevention
From bolstering patient engagement to navigating ethical dilemmas, these modules ensure you’re well-versed in managing participant interactions. Learn how to identify motivating factors for recruitment, address compliance challenges, and recognize red flags for misconduct or fraud.
Regulatory Mastery with TMFs and CFR 21 Part 11
Managing Trial Master Files (TMFs), essential regulatory documents, and electronic signatures per CFR 21 Part 11 is simplified with these practical modules. The emphasis on data accuracy and reliability ensures that no detail is overlooked.
Final Preparations and Certification
The program concludes with real-world exercises and an exhaustive evaluation through case studies and a certification exam. By the end, you’ll be fully equipped to excel in advanced monitoring practices and meet the demands of a senior-level CRA.
Why Choose the ACRAC Syllabus?
The ACRAC syllabus sets itself apart with its depth, versatility, and applicability. With practical insights, global relevance, and accreditation from esteemed institutions, it’s not just a certification—it’s a career-transforming experience.
Start Your Journey Today
Whether you’re beginning your career or looking to level up, the ACRAC program gives you the tools and training to thrive in clinical research. Explore the program and enroll now at CCRPS ACRAC to start advancing your skills. With these 196 modules behind you, success is just around the corner!

Get ahead in clinical research with advanced accredited online CRA certification for $450. Demo our on-demand course below.
Clinical Research Associate Certification
References
Beroe Inc. - Explore the latest insights and market intelligence on clinical research organizations, focusing on trends, challenges, and opportunities in the industry. Visit Beroe Inc.LinkedIn Jobs - Discover entry-level Clinical Research Associate positions across the United States, leveraging your network to find new opportunities. Explore LinkedIn Jobs
CenterWatch - Read about the ongoing shortage of qualified CRAs and its impact on the pharmaceutical industry, including increased costs and project delays. Read more on CenterWatch
NCBI - Access a comprehensive article on clinical research, providing valuable insights into methodologies and best practices. Read the article on NCBI
NIAID - Learn about the DMID's role in investigational product research, including guidelines and protocols. Visit NIAID
FDA - Understand the different types of clinical research and their significance in advancing medical knowledge. Explore FDA's guide
Dixon JR. 1999 - Review the international conference on harmonization good clinical practice guideline, focusing on quality assurance in clinical trials. DOI: 10.1080/105294199277860
FDA E6(R2) Guidelines - Review the comprehensive guidelines for Good Clinical Practice, ensuring compliance and ethical standards in clinical trials. Access the guidelines
WHO - Discover the recommended format for research protocols as outlined by the WHO's Research Ethics Review Committee. Visit WHO
IAOCR - Find guidance on securing your first clinical research job, with tips and strategies for breaking into the field. Explore IAOCR
New Scientist - Gain insights into building a career in clinical research, including necessary skills and industry trends. Read more on New Scientist
NCBI - Delve into data management practices in clinical trials, focusing on efficiency and accuracy. Read the article on NCBI
St. Germain DC, Good MJ. 2017 - Explore data management in clinical trials, as detailed in the book "Principles and Practice of Clinical Research." ISBN 978-0-12-849905-4
ClinEssentials - Offers a variety of courses, products, and services designed to help clinical research professionals thrive and achieve fulfilling careers. Visit ClinEssentials
Clinical Leader - Explore insights and advice for starting a career in clinical research, including common challenges and solutions. Read more on Clinical Leader
ProClinical - Learn how to secure a job as a Clinical Research Associate, with tips on standing out in the application process. Visit ProClinical
SWOG Clinical Research Resources - Provides a comprehensive collection of links to useful resources related to clinical trials, including those from the FDA, OHRP, OCR, NIH, and NCI. Explore SWOG Resources
College Choice - Review the best master's degree programs in clinical research, helping you choose the right path for advanced education. Visit College Choice
ClinEssentials - Offers a variety of courses, products, and services designed to help clinical research professionals thrive and achieve fulfilling careers. Visit ClinEssentials
SWOG Clinical Research Resources - Provides a comprehensive collection of links to useful resources related to clinical trials, including those from the FDA, OHRP, OCR, NIH, and NCI. Explore SWOG Resources

Clinical Research Courses - CTA vs. CRC vs. CRA
The Ultimate Guide to Clinical Research Courses for CTA, CRA, and CRC Professionals
What Clinical Research Career and Course Is Best for You? Take Career Quiz
Transform Your Future with a Career in Clinical Research
Clinical research is one of the most exciting fields in healthcare, offering endless opportunities to make a real impact while building a fulfilling and rewarding career. Whether you’re beginning your journey as a Clinical Trial Assistant (CTA), preparing to step up as a Clinical Research Associate (CRA), or considering a pivotal role as a Clinical Research Coordinator (CRC), the key to success is understanding the qualifications, experience, and training needed for each role.
Start Here: Is CTA, CRA, or CRC Right for You?
Courses
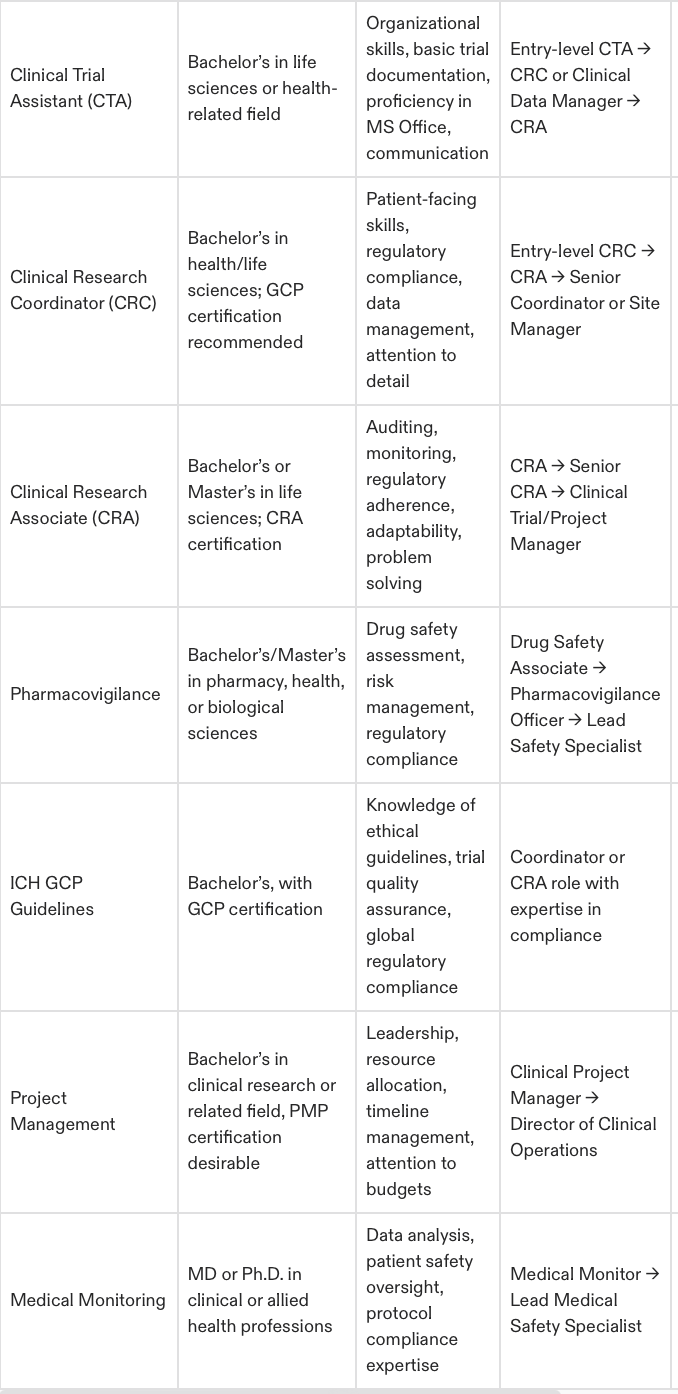
1. Clinical Trial Assistant (CTA)
What Does a CTA Do?
CTAs provide essential support to clinical trials, ensuring all administrative and operational tasks are handled efficiently. They are the backbone of trial documentation and site communication efforts, making them a vital part of the research process.
Background & Education
Ideal Background: Exceptional organizational skills and a keen eye for detail. Many CTAs come from administrative, healthcare, or life sciences-related roles.
Education Required: Most employers look for a bachelor’s degree in life sciences or health sciences. If you don’t have a degree, strong administrative experience in healthcare or laboratory settings may qualify you.
Experience and Career Path
You don't need prior experience to get started as a CTA. Many CTAs gain on-the-job training, providing them with foundational knowledge of clinical trial operations.
Career Advancements:
Start as a CTA managing trial documentation and logistics.
Transition into roles like Clinical Research Coordinator (CRC) or Clinical Data Manager.
Move up to CRA positions with further training and experience.
CTA to CRC – The Logical Career Step Forward
For CTAs looking to advance quickly, moving into a CRC role offers better earning potential, increased responsibilities, and hands-on participation in clinical trials. The CCRPS CRC Course can enable this transition within weeks.
2. Clinical Research Coordinator (CRC)
What Makes a CRC Different?
CRCs are essential players in clinical trials, ensuring that studies run smoothly at the site level. They oversee patient enrollment, documentation, and compliance while working closely with investigators and patients. This role provides more meaningful interaction within clinical research and serves as a stepping stone to higher-responsibility roles like CRA.
Background & Education
Ideal Background: Candidates from healthcare, nursing, or administrative roles often transition seamlessly into CRC positions.
Education Required: A bachelor’s degree in life sciences, health sciences, or related fields is generally required. Training in Good Clinical Practice (GCP) and human subjects protection will also set you apart.
Experience and Career Path
Experience requirements vary, but a foundation in clinical administration or exposure to patient care is advantageous. Many CRCs start as CTAs or healthcare workers and leverage training to secure their position.
Career Advancements:
Begin as a CTA or similar entry-level role.
Transition into a CRC role with certifications like those offered by CCRPS.
Progress to leadership or CRA roles with site management experience.
For faster career growth, the CCRPS CRC Course equips you with the skills and certification recognized by industry leaders.
3. Clinical Research Associate (CRA)
What to Expect as a CRA
CRAs hold one of the most respected and vital roles in clinical research. They are responsible for monitoring clinical trials, ensuring compliance with regulatory standards, verifying data accuracy, and protecting patient safety.
Background & Education
Ideal Background: Many CRAs come from CRC or healthcare roles, transitioning into monitoring positions.
Education Required: A bachelor’s or master’s degree in life sciences, health sciences, or a related field is required. Candidates with advanced degrees (e.g., PhDs) are also highly sought after, especially for specialized therapeutic areas.
Experience and Career Path
CRA positions typically require 1–3 years of experience in clinical trial coordination, site management, or monitoring. Most professionals advance from CRC roles or pursue specialized CRA training.
Career Advancements:
Start as a CRC, gaining site-specific knowledge.
Transition into CRA monitoring roles with comprehensive training.
Further your career as a Clinical Trial Manager (CTM) or Director of Clinical Operations.
Why Training is Key for CRA Success
With extensive responsibilities requiring in-depth knowledge, CRA certifications like the CCRPS CRA Course are indispensable in preparing for this challenging yet rewarding role.
Career Comparison

Clinical Research Courses
Why Choose CCRPS for Your Training?
At CCRPS, we specialize in providing accessible, comprehensive, and industry-approved training programs to advance your clinical research career.
Tailored Training Programs
Learn everything from trial documentation to AE/SAE handling, regulatory compliance, and data management.
Flexibility for Busy Schedules
Study at your own pace with affordable programs that can be completed in just 2–4 weeks.
Proven Success
With over 8 years of graduate success, CCRPS courses have empowered thousands of professionals to reach their career goals.
Affordable Tuition with Big Results
Quality education shouldn’t break the bank—CCRPS ensures value for your investment.
Explore Our Industry-Aligned Training Programs:
Ultimate Guide to Selecting Clinical Research Courses
Why Clinical Research Training Matters
Clinical research bridges the gap between medical innovation and patient care, ensuring new treatments are both safe and effective. If you’re eager to build a career in this dynamic field, start by choosing the right training. Whether you’re aiming to become a Clinical Trial Assistant (CTA), Clinical Research Coordinator (CRC), or Clinical Research Associate (CRA), the course you select will directly impact your knowledge, skills, and career trajectory.
This guide offers an in-depth approach to selecting the best clinical research course, including practical tips and an overview of why CCRPS is a trusted leader in clinical research training.
Factors to Consider When Selecting a Clinical Research Course
To find the program that matches your needs, focus on these critical aspects when evaluating clinical research courses.
1. Course Content That Matches Your Goals
A robust curriculum is the backbone of a reliable clinical research course. Assess whether the program includes the following key topics:
Good Clinical Practices (GCP)
Protocol development and trial management
Patient safety, informed consent processes, and Adverse Event (AE/SAE) reporting
Regulatory compliance and global standards (e.g., FDA, EMA)
Trial documentation, data capture, and site management
For advanced roles like CRC or CRA, look for specialized modules. For example, CCRPS provides separate programs tailored to the unique requirements of CRCs, CRAs, and CTAs, ensuring you’re prepared for the specific challenges of your role.
Tip: Request a syllabus or detailed breakdown of the course content before you enroll to verify alignment with your career path.
2. Accreditation and Industry Recognition
A course’s accreditation speaks volumes about its quality and credibility. Choose programs endorsed by reputable organizations or recognized by employers in the clinical research field. Accreditation ensures that the course meets rigorous standards and carries weight with hiring managers.
CCRPS courses are widely accepted across the industry and designed to meet global standards, giving graduates an advantage in their job search.
3. Flexibility and Accessibility
Your training shouldn’t interfere with your current commitments. Look for courses that offer:
Online learning for remote accessibility.
Self-paced schedules to accommodate your busy life.
User-friendly platforms that allow you to study from any device.
CCRPS courses are 100% online, self-paced, and accessible 24/7, allowing you to learn whenever it’s convenient for you.
4. Cost and Affordability
While education is essential, staying within budget matters. Evaluate the total cost of the course, including hidden fees, materials, and certification. A program should strike a balance between cost-effectiveness and quality of education.
CCRPS is renowned for offering high-quality courses at competitive prices while ensuring transparency in pricing. You get exceptional value without compromising on content.
5. Practical Learning Opportunities
While theory forms the foundation, practical application of knowledge is key to excelling in clinical research roles. Opt for courses that emphasize hands-on learning through:
Case studies and real-world examples.
Exposure to tools like Clinical Trial Management Systems (CTMS).
Scenarios that mimic clinical trial environments.
CCRPS incorporates practical, job-ready learning components to give students a real sense of what working in clinical research entails.
6. Career Outcomes and Employer Connections
Ultimately, your course should prepare you for the next step in your career. Evaluate the program's success in helping graduates secure roles. Check:
Job placement statistics.
Graduate feedback and testimonials.
Resources for resume building and career support.
CCRPS boasts a 90%+ job placement success rate and provides tailored resources like career counseling and employer-recognized certifications to set you apart.
7. Provider Reputation
Look for course providers with a proven track record of training success. Trusted providers, like CCRPS, not only offer excellent content but also provide ongoing support and maintain their standing within the clinical research industry.
Tip: Use reviews, ratings, and alumni testimonials to get a sense of how graduates feel about the program and its real-world application.
Why CCRPS is the Best Choice for Clinical Research Training
When it comes to clinical research education, CCRPS stands out as a comprehensive, student-focused provider. Here’s what makes it the best choice for your training needs:
1. Specialized Curriculum to Match Career Goals
CCRPS offers targeted CTA, CRC, and CRA courses that ensure you train specifically for your desired role. Their curriculum is industry-designed and regularly updated to meet employer standards.
2. Flexible and Fully Online Training
With CCRPS, you can study on your own schedule, anywhere in the world. This accessibility means professionals with busy lives don’t have to sacrifice convenience for quality.
3. Proven Career Placement Success
CCRPS certifications are trusted by sponsors, CROs, and research institutions globally. Most graduates report significant career advancement or job offers shortly after completing their training.
4. Affordable Pricing with Excellent Value
CCRPS strives to make quality education affordable. You’ll gain access to industry-standard training without overspending.
5. Practical Learning for Real-World Success
CCRPS blends theory with hands-on learning through case studies and real-world scenarios designed to mirror clinical trial challenges.
6. Career Support
From resume reviews to personalized career counseling, CCRPS ensures graduates feel prepared to enter the workforce with confidence.
Final Checklist to Choose the Right Course
Before making your decision, answer these questions using the information provided by the course provider:
Does the curriculum meet my career goals?
Is the course accredited or widely recognized?
Is the course flexible and accessible for my schedule?
Is the pricing clear and affordable?
Do graduates have proven success in advancing their careers?
If the answer to all these questions is "yes," you’re on the right track. CCRPS ticks every box and has a proven record of producing industry-ready professionals.
Your Clinical Research Career Starts Here
Selecting the best clinical research course is the first step toward a rewarding future in this impactful field. With CCRPS, you’ll gain the training, certifications, and confidence needed to succeed and grow in this dynamic industry.
Explore programs tailored to your goals now:
Your Career in Clinical Research Starts Now
The clinical research industry offers incredible growth potential for professionals who invest in the right training. Whether you’re beginning your career as a CTA, transitioning to a CRC, or striving to become a CRA, CCRPS is here to help you achieve your goals.
Take control of your future today with CCRPS’s comprehensive training programs—your gateway to financial stability, career growth, and making a real difference in healthcare.
Enroll now and start building your clinical research career today:
What Clinical Research Course Is Best for You? Take Career Quiz






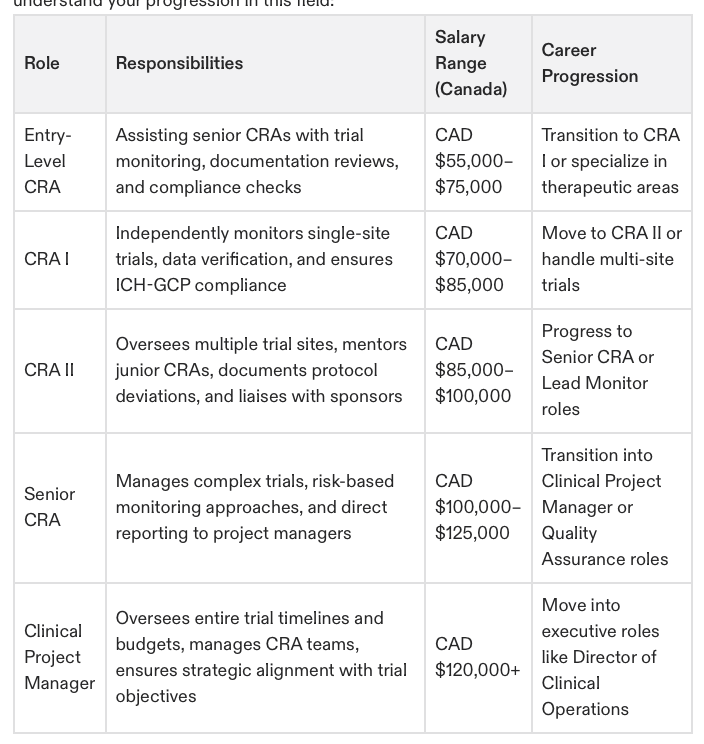
The Ultimate Guide to Becoming a Clinical Research Associate (CRA) in Canada: Everything You Need to Know in 2025
Clinical Research Associate - For Canada
If you’ve been exploring healthcare or clinical research careers, you might have stumbled upon the exciting opportunity of becoming a Clinical Research Associate (CRA). The clinical research associate career not only offers a dynamic work environment but also provides lucrative career prospects in a fast-growing field. Whether you’re in Canada or looking to elevate your career globally, this ultimate guide will outline everything you need to know and reveal how the Certified Clinical Research Professionals Society (CCRPS) course can fast-track your success.
By the end of this guide, you’ll have the tools, strategies, and resources to master the CRA path while maximizing your career potential.
What Is a Clinical Research Associate (CRA): The Role Explained
A Clinical Research Associate is at the heart of clinical trials. CRAs are responsible for clinical monitoring to ensure they comply with strict ethical, scientific, and regulatory requirements. They are often called the “eyes and ears” of clinical trials, as they oversee site operations, data accuracy, and compliance.
Key Responsibilities:
Monitoring trial sites for protocol adherence and data accuracy.
Ensuring that the trial complies with Good Clinical Practice (GCP) guidelines.
Validating collected data and resolving discrepancies.
Acting as a liaison between trial sponsors and site investigators.
Training site staff on compliance and reporting procedures.
CRAs travel frequently—or work remotely in the case of decentralized trials—making it a dynamic career choice for those who enjoy a mix of structure and flexibility.
Clinical Research Associate Job Description and Skills
A Clinical Research Associate (CRA) plays a crucial role in the clinical research process, ensuring that clinical trials are conducted in accordance with good clinical practice guidelines and regulatory requirements. The primary responsibilities of a CRA include:
Monitoring clinical trials to ensure compliance with the clinical trial protocol, good clinical practice guidelines, and regulatory requirements.
Conducting site visits to assess the quality of data and ensure that the clinical trial is being conducted in accordance with the protocol.
Reviewing and verifying case report forms (CRFs) to ensure that data is accurate and complete.
Identifying and addressing issues that may impact the quality of the clinical trial.
Collaborating with clinical research coordinators, investigators, and other stakeholders to ensure that the clinical trial is conducted efficiently and effectively.
To be successful in this role, a CRA must possess a range of skills, including:
Strong knowledge of good clinical practice guidelines and regulatory requirements: Understanding these guidelines is essential for ensuring that clinical trials are conducted ethically and effectively.
Excellent communication and interpersonal skills: CRAs must be able to communicate effectively with site staff, investigators, and sponsors.
Ability to work independently and as part of a team: CRAs often work autonomously but must also collaborate with various stakeholders.
Strong organizational and time management skills: Managing multiple tasks and responsibilities efficiently is crucial.
Ability to analyze data and identify trends and issues: CRAs need to be detail-oriented and capable of spotting discrepancies in clinical trial data.
Strong attention to detail and ability to maintain accurate records: Precision is key in ensuring the integrity of clinical trial data.
Ability to work in a fast-paced environment and prioritize multiple tasks: The dynamic nature of clinical trials requires adaptability and effective prioritization.
In addition to these skills, a CRA must also have a strong understanding of clinical research principles, including the design and conduct of clinical trials, data management, and statistical analysis.
Career Pathway
Entry-Level Roles such as Clinical Research Coordinator (CRC) or Clinical Trial Assistant (CTA). Clinical researchers play a crucial role in training and mentoring entry-level CRAs.
CRA I/II Levels focus on independent monitoring and cross-site management.
Senior CRA or Lead CRA positions involve mentoring or managing multiple trial teams.
Career growth can also include moving into Clinical Project Manager roles, where salaries often exceed six figures.
Salary Expectations: Entry-level CRAs in Canada earn approximately CAD $55,000 to $75,000, with senior roles reaching CAD $120,000 or more.
Step 1: Educational Requirements to Become a CRA
Minimum Academic Credentials
Most CRA positions require at least a bachelor’s degree in a life science discipline (e.g., Biology, Biochemistry, Pharmacy, or Nursing). Advanced degrees, such as a Master’s in Clinical Research, provide added advantages but are not mandatory for starting.
For non-traditional candidates, certification programs like CCRPS (Certified Clinical Research Professionals Society)bridge knowledge gaps and prepare individuals for entry-level CRA roles, even without prior research experience. These programs also ensure that individuals are equipped to adhere to rigorous standards in clinical research studies.
Pro Tip: Canadian universities like McMaster University or York University offer specific programs in clinical trial management, blending academic learning with hands-on training.
Step Up With Certifications
Certifications enable faster entry into the CRA field and elevate your marketability. The CCRPS course is particularly advantageous as it offers a thorough curriculum across 165 modules, mentorship support, and job placement resources.
Certification Program
To determine which program suits your goals and experience, here’s a quick comparison of major CRA certification programs:
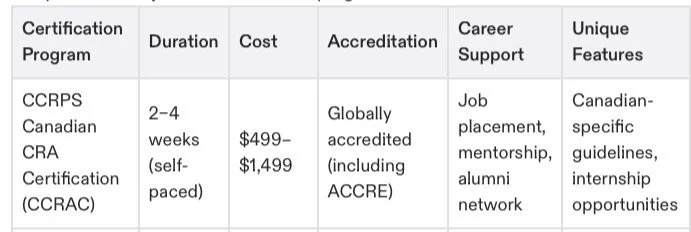
Canadian CRA Certification
Note: CCRPS’s flexibility and comprehensive curriculum make it an excellent choice for both beginners and those transitioning from other fields.
Step 2: Why the CCRPS Course Is a Game-Changer for Your Clinical Research Associate Career
The Certified Clinical Research Professionals Society (CCRPS) provides one of the most comprehensive CRA training programs aimed at both beginners and experienced professionals.
Here’s why it's the go-to certification for boosting CRA prospects in Canada or internationally:
Globally recognized certification suitable for beginners and advanced professionals.
Offers flexible payment plans and a 100% money-back guarantee.
Includes advanced courses covering specific therapeutic areas, such as pediatric and oncology trials.
CCRPS also provides mentorship opportunities and a career-building alumni network. These exclusive benefits ensure graduates are better positioned to land higher-paying roles.
Step 3: CRA Career Levels and Responsibilities
Here’s a detailed comparison of CRA roles at various career levels to help you understand your progression in this field. Many CRAs find employment opportunities within contract research organizations (CROs), which conduct clinical trials and provide essential resources for pharmaceutical firms:
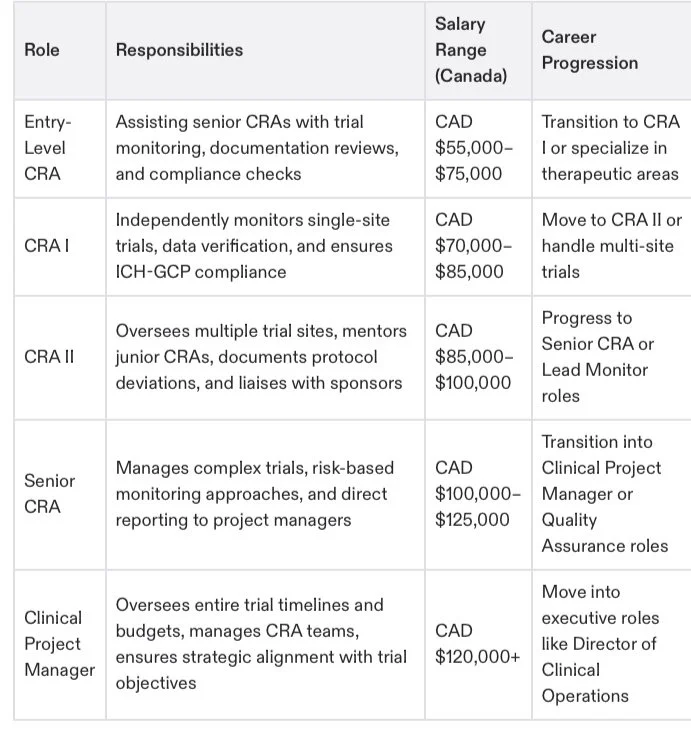
CRA Career Ladder
Note: Specialized certifications like CCRPS can expedite progression to higher roles by equipping CRAs with advanced, niche monitoring capabilities.
Step 4: Mastering Advanced Monitoring Techniques in the Clinical Research Process
The CCRPS course gives CRAs a competitive edge by offering specialized modules in risk-based monitoring, patient retention, and regulatory compliance for complex studies. Graduates often report accelerated promotions and salary growth due to these advanced skills.
Tools to Master Through CCRPS Training for Clinical Trials:
Electronic Data Capture (EDC) systems such as Medidata or Oracle Clinical.
Trial Master File (TMF) processes for streamlined documentation.
Risk-Based Monitoring Platforms adopted by leading CROs.
How to Get Experience in Clinical Research
Gaining experience in clinical research is a critical step towards becoming a successful Clinical Research Associate. Here are several ways to build your experience:
Volunteering or interning at a research institution or hospital: This provides hands-on experience and exposure to the clinical research environment.
Participating in clinical trials as a research coordinator or assistant: These roles offer practical experience in managing and monitoring clinical trials.
Taking courses or earning a degree in a field related to clinical research, such as life sciences or public health: Formal education provides a strong foundation in the principles of clinical research.
Joining professional organizations, such as the Association of Clinical Research Professionals (ACRP) or the Society of Clinical Research Associates (SOCRA): These organizations offer networking opportunities, resources, and professional development.
Networking with experienced clinical research professionals and seeking their advice and guidance: Building relationships with industry professionals can provide valuable insights and career opportunities.
Participating in online forums and discussion groups related to clinical research: Engaging in these communities can help you stay informed about industry trends and best practices.
It’s also important to note that many clinical research positions require a degree in a field related to life sciences or public health, as well as relevant experience. However, there are many entry-level positions available for those who are new to the field, providing a pathway to gain the necessary experience and advance in your career.
Understanding ICH-GCP Guidelines
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Good Clinical Practice (GCP) guidelines are a set of principles and guidelines that ensure clinical trials are conducted in a way that is safe, efficient, and effective. These guidelines cover a range of topics, including:
The design and conduct of clinical trials: Ensuring that trials are scientifically sound and ethically conducted.
The protection of human subjects: Safeguarding the rights, safety, and well-being of trial participants.
The collection and management of data: Ensuring data integrity and accuracy throughout the clinical trial process.
The monitoring and auditing of clinical trials: Regular oversight to ensure compliance with the trial protocol and regulatory requirements.
The reporting of adverse events and other safety issues: Prompt and accurate reporting of any issues that may affect participant safety.
The ICH-GCP guidelines are widely accepted and used by regulatory agencies around the world to ensure that clinical trials are conducted in accordance with good clinical practice. Clinical research professionals must have a strong understanding of these guidelines and be able to apply them in their work.
In addition to the ICH-GCP guidelines, clinical research professionals must also be familiar with other regulatory requirements, such as those related to electronic data capture and the use of technology in clinical research. Understanding these guidelines and requirements is essential for ensuring the integrity and success of clinical trials.
What Does a Clinical Research Associate (CRA) Do?—A Complete Role Breakdown
A Clinical Research Associate acts as the key guardian of clinical trials, ensuring compliance with regulatory and ethical frameworks. Functioning as the "eyes and ears" of trial sponsors, CRAs validate that trials operate according to Good Clinical Practice (GCP) guidelines while delivering reliable data aligned with global safety standards.
Core CRA Responsibilities
Monitoring Trial Sites: Ensure trial teams follow trial protocols and safeguard participant safety.
Data Verification: Match source documents with entries in electronic data systems like CTMS or Medidata.
Regulatory Adherence: Confirm that sites meet International Council for Harmonisation (ICH) GCP standards.
Site Training: Prepare research teams through hands-on training to maintain compliance.
Problem Resolution: Resolve deviations, errors, or logistical challenges found within trial processes.
Emerging Trends in CRA Roles for 2024
Decentralized and Hybrid Trials leveraging remote monitoring platforms like eTMF and tech-driven analytics.
Specialized CRAs focusing on therapeutic niches like oncology, gene therapy, or pediatric trials.
The rise of Risk-Based Monitoring (RBM) emphasizing targeted tracking of high-priority sites and reducing redundant oversight.
Pro Tip: Build expertise with tools like Oracle Clinical, Medidata RAVE, and Veeva Vault to accelerate entry.
Did You Know? CRAs are well-compensated for their critical roles. Salaries for entry-level CRAs begin at USD $55,000 (CAD $70,000 in Canada), and experienced professionals earn upwards of USD $120,000.
How to Become a Clinical Research Associate—Your Career Blueprint
Succeeding in clinical research doesn’t happen overnight. Follow these structured steps to create a strong foundation for this exciting role.
Step 1. Obtain a Relevant Degree or Equivalent Experience
To qualify as a CRA, most hiring managers prioritize candidates with at least a bachelor’s degree in these fields:
Life Sciences (Biology, Biochemistry, Genetics)
Medicine or Nursing
Pharmacy or Biotechnology
Unlike some rigid healthcare careers, prospective CRAs can also transition from roles like clinical trial assistants or healthcare data analysts.
Considerations for Canadian CRAs
Canadian CRAs should familiarize themselves with Health Canada’s regulatory guidelines in addition to global compliance systems like FDA requirements. Institutions like McMaster University offer specific clinical trial management courses tailored for Canadians.
Step 2. Build Initial Exposure Through Entry-Level Roles
Before securing a CRA role, gaining hands-on experience significantly enhances applications. Pursue:
Clinical Research Coordinator (CRC) roles handling day-to-day site management.
Administrative positions in medical research hubs like Toronto General Hospital or CRA internship programs.
Leverage volunteer programs with hours-based work to understand real-world settings.
Key Skills and Tools for a CRA
This table provides a detailed comparison of the key skills and tools CRAs need to perform their role effectively.
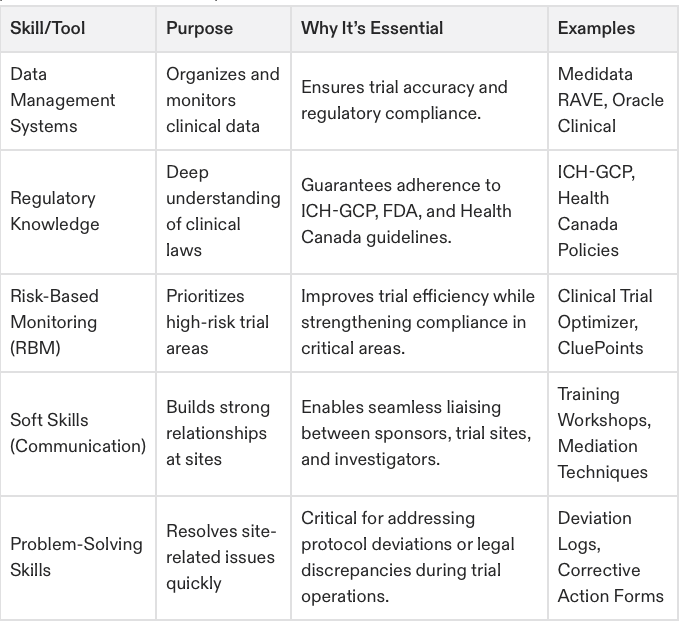
Section 1. Regulatory Compliance Excellence
Regulatory compliance is the backbone of a successful CRA career. CRAs must ensure that every aspect of a clinical trial aligns with ethical and legal standards.
Core Compliance Concepts
ICH-GCP Guidelines: A universal standard for clinical trials, ensuring participant safety and scientifically sound results.
Health Canada Regulations: Canadian CRAs must follow federal policies concerning trials conducted on Canadian soil.
FDA Compliance (for U.S.-run trials): Required if working on global or U.S.-based projects.
Pro Tip: Regularly review and stay updated on changing regulations. CCRPS modules include exclusive content on international and Canadian-specific compliance.
Section 2. Effective Communication Strategies for CRAs
For CRAs, communication is not optional—it’s a decisive factor in trial success. Whether training site staff, interacting with sponsors, or resolving issues, mastering communication ensures clarity.
How to Communicate Effectively as a CRA
Be Proactive: Schedule regular check-ins with study sites to preempt potential protocol deviations.
Tailor Messages: Adapt tone and detail based on your audience, whether site managers, sponsors, or healthcare professionals.
Use Best Practices in Documentation: Every CRA should prioritize error-free, comprehensive communication logs for trial traceability.
Tool to Try: Try role-based simulations through the CCRPS Fellowship Program to sharpen both verbal and electronic communication.
Section 3. Career Advancement Tips for CRAs
Whether you’re just starting or seeking to climb the career ladder, follow these tips to accelerate your progress in clinical research.
Get Certified: Ensure your resume reflects globally recognized credentials like CCRPS certification and the CRAC credentialing exam (Canada).
Specialize in Therapeutic Areas: Advanced certifications in oncology or cardiovascular trials improve your standing within niche fields.
Broaden Networking Opportunities: Join CRA-specific organizations, such as the Canadian Clinical Trials Network, or attend industry events to discover hidden job opportunities.
Key Points on Excelling as a Clinical Research Associate
Mastering the CRA role requires more than technical knowledge—you need strategic actions, strong interpersonal skills, and certification. By following this guide’s recommendations, CRA candidates worldwide, particularly in Canada, can confidently pursue fulfilling and lucrative careers.
From enhanced certifications like CCRAC to advanced tools like RBM tracking systems, being innovative in your career opens avenues for accelerated growth. Start where you are, get certified, and watch your position in the clinical research world evolve into a leading role.
Get started today on your Canadian CRA career!
Enroll in CCRPS to Begin Your Journey
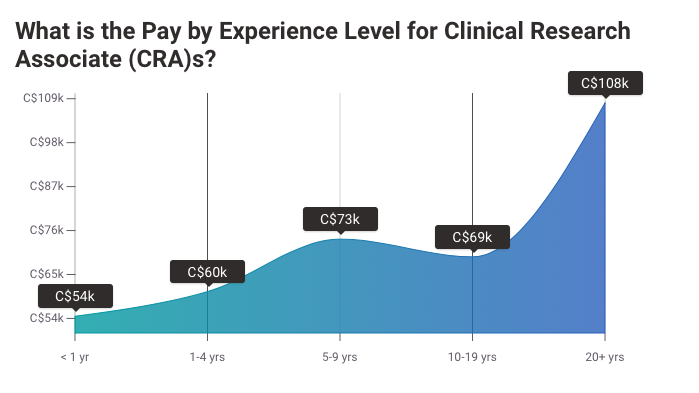
Payscale Canadian CRA Salary

Payscale Canadian Pay Difference CRA
Clinical Research Certification Specifically for Canadian CRAs
CCRPS offers one of the only online certifications specifically designed for entry-level and mid-level professionals in clinical research associate knowledge. We prepare you to understand and apply all skills required to secure a job in clinical research.
Want to Learn More About Clinical Research?
Take courses from CCRPS and learn more on how to become a clinical research professional.
Discover more from Clinical Research Training | Certified Clinical Research Professionals Course
Discover more from Clinical Research Training | Certified Clinical Research Professionals Course








Clinical Trial Coordinator: Guide to the Roles and Duties of a Clinical Trial Coordinator
A Clinical Trial Coordinator (CRC) is essential to the success of any clinical trial. Tasked with managing research activities at clinical sites, CRCs ensure compliance with protocols, Good Clinical Practice (GCP) guidelines, and regulatory requirements. These professionals bridge the gap between key stakeholders, including sponsors, Ethics Committees, and investigators, while prioritizing the integrity of trial processes and the safety of participants.
The role is multifaceted—combining organization, technical know-how, ethical judgment, and a strong commitment to excellence. Given the criticality of their functions, comprehensive training, such as CRC certification, is indispensable for excelling in this field. Below, we offer a detailed exploration of a CRC’s roles, responsibilities, tools, and opportunities for growth.
CRC Responsibilities Across Trial Phases
The responsibilities of a CRC span distinct trial phases, each requiring unique skills and attention to detail. By breaking these down, we can illustrate the diverse nature of their work:
1. Pre-Trial Responsibilities

Before a clinical trial begins, the CRC plays a crucial role in ensuring the research site is ready to meet ethical, regulatory, and operational standards. This phase involves extensive documentation, liaising with key stakeholders, and managing logistics.
Key Tasks Include:
Study Feasibility Assessments:
Oversee completion of questionnaires submitted by sponsors or CROs to determine the site’s suitability.
Provide logistics details (e.g., investigator credentials, infrastructure capacity).
Assist in finalizing the site selection process through pre-study site visits.
Ethical and Regulatory Preparation:
Collate and submit documentation for Ethics Committee approval, which includes the investigator CVs, study protocols, insurance certifications, and patient diaries.
Schedule investigator meetings to align all parties and ensure the site is prepared for trial initiation.
Training and Readiness:
Conduct onboarding for site staff to ensure familiarity with trial requirements.
File documents such as signed protocols, blank CRFs, and clinical trial coordinator certifications.
Example Scenario:
Considering a study testing a novel cancer drug, a CRC may work alongside oncologists to ensure the site meets advanced treatment administration capabilities, procure legal documentation, and carry out pre-site inspection visits with CRO representatives.
2. Responsibilities During the Trial
Once the trial starts, the CRC moves into an operational phase. This involves managing participants, ensuring data accuracy, and maintaining compliance with the protocol.

Key Tasks Include:
Participant Recruitment and Management:
Screen potential participants against inclusion and exclusion criteria.
Obtain informed consent, ensuring participants fully understand the trial procedures and risks.
Schedule visits and coordinate participant timelines to align with protocol requirements.
Data Oversight:
Use tools like IVRS/IWRS to randomize participants and document visit information.
Maintain detailed case report forms (CRFs) and ensure prompt submission to sponsors or Data Monitoring Committees (DMC).
Monitoring Adverse Events:
Record adverse events (AEs) and serious adverse events (SAEs), carefully documenting details such as drug dosage, administration route, and reactions.
Communicate all safety concerns to the Principal Investigator (PI) and sponsor for review and action.
Managing Medications and Equipment:
Oversee the storage and management of investigational products (IPs), ensuring compliance with pre-defined temperature and safety protocols.
Collaborate with site pharmacists to maintain drug accountability logs.
Example Scenario:
For a clinical trial involving wearable health monitors, a CRC may train participants on proper use, troubleshoot device issues, and ensure collected data aligns with the study protocol.
3. Post-Trial Responsibilities
The CRC’s work doesn’t end when the last participant visit is completed. The close-out phase is equally vital to ensure the trial meets archival and auditing standards.
Key Tasks Include:
Trial Close-Out:
Assist sponsors and CRAs during site close-out visits, resolving any documentation discrepancies.
Review and organize trial data in preparation for audits.
Document Archival:
Prepare study documents for long-term storage, ensuring regulatory compliance for retention periods (often 15-20 years).
Maintain updated inventories of trial documentation to respond swiftly to inquiries during follow-ups.
Additional Areas of Impact
Beyond the standard phases, CRCs have roles that require understanding of communication, technology, and industry trends.
Communication Skills and Stakeholder Management
Effective communication is at the core of a CRC’s responsibilities. They serve as a bridge between investigators, site staff, sponsors, and Ethics Committees.
Internal Team Coordination: CRCs foster collaboration across the clinical site team to ensure smooth implementation of the trial protocol.
Sponsor Relationships: These professionals maintain regular reporting with sponsors or CROs, offering updates on site progress and addressing challenges.
Example:
If a trial suddenly requires re-negotiating the clinical research agreement, a CRC’s communication skills will support seamless adjustments without delaying the trial.
Technological Tools in Clinical Research
Technology is integral for efficient trial management. CRCs use various tools to monitor compliance, record data, and enhance trial operations.
Data Management Tools: Electronic Data Capture (EDC) systems like REDCap or Medidata streamline documentation and reporting.
Participant Support: Telehealth tools expand recruitment efforts and assist with remote monitoring in decentralized trials.
Ethics and Problem-Solving
CRCs regularly encounter situations requiring sound ethical judgment. Whether managing protocol deviations or ensuring participant well-being, their decision-making has a lasting impact on trial success.
Example:
If a rural participant faces transportation challenges, a CRC may work with sponsors to arrange alternative solutions, such as remote assessments.
Career Opportunities and Certification Advantage
The demand for Clinical Trial Coordinators is increasing alongside the global growth of clinical trials. Certification offers multiple benefits, including:
Enhanced Skill Validation: CRC certification demonstrates expertise in trial management.
Career Progression: Suitable roles include Clinical Research Associates (CRAs), Project Managers, or Regulatory Specialists.
Global Opportunities: Proficiency in GCP standards enables CRCs to work internationally, adapting to diverse regulatory environments.
Why Choose CCRPS for Certification?

At CCRPS, we offer tailored certification programs to prepare you for success in clinical research.
Comprehensive Training: From CRC certification to ICH GCP training, our courses focus on critical skills such as ethical considerations, protocol implementation, and regulatory compliance.
Flexibility and Accessibility: Self-paced modules allow you to balance training with ongoing work.
Advanced Expertise Options: Explore Advanced Clinical Project Manager Certification or Principal Investigator Certification for accelerated career growth.
Recommended Courses:
Clinical Research Coordinator Certification: Ideal for aspiring CRCs looking to master trial management.
Pharmacovigilance Training: Deepen your understanding of drug safety and adverse events monitoring.
CRA Training: Perfect for CRCs seeking a transition into a monitoring role.
Take Action Today
Empower your career with advanced training from CCRPS. Whether you're starting in clinical research or aiming for career advancement, our certifications provide the edge you need.
Start your CRC certification today!
What Is ICH GCP Certification 2024
What Is ICH GCP Certification?
ICH, which stands for the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, is a pivotal global organization shaping the landscape of clinical trials in 2024. Responsible for setting stringent standards, ICH ensures that clinical trials involving human subjects adhere to rigorous regulatory frameworks.
In essence, ICH oversees the implementation of Good Clinical Practice (GCP) guidelines, ensuring that the planning, execution, and documentation of clinical trials are conducted ethically and with scientific integrity at every phase of the research process.
For those seeking to enhance their understanding of ICH GCP guidelines, there's an opportunity to access free online training, providing invaluable insights into the intricacies of conducting ethical and scientifically sound clinical trials in 2024.
You can demo ich gcp training free online here.
What is the purpose of GCP Certification?
In 2024, GCP Certification, short for Good Clinical Practice Certification, serves as a crucial acknowledgment of an individual's proficiency and expertise in implementing regulatory guidelines across all facets of their work.
Why is it significant?
In the realm of clinical research, possessing GCP certification is indispensable for success. It's not just a matter of preference; it's a necessity. Any organization involved in clinical research endeavors must ensure that their personnel comprehensively grasp the ICH guidelines and are certified to conduct scientific studies accordingly. In fact, many companies provide certification programs for their employees even before they commence their duties. This underscores the paramount importance of GCP certification in the clinical research industry landscape of 2024.ICH GCP Attestation Form
If you're looking to explore a career in the innovative and quickly growing field of clinical research, you'll need to ensure you get the proper Good Clinical Practice certification.
GCP Certification
Are you seeking a GCP refresher online course or comprehensive initial advanced GCP training in 2024? Look no further. Our cutting-edge GCP certification courses are designed to equip you with all the necessary tools to obtain your certification efficiently.
In today's fast-paced world, attending in-person classes for GCP certification training may not always be feasible. That's why our innovative and user-friendly online courses offer a convenient alternative. We understand the importance of receiving top-notch training from experts well-versed in the ICH GCP guidelines to propel your career forward.
At CCRPS, excellence is our standard. Each ICH GCP module we offer is crafted with meticulous attention to detail, ensuring that you receive the highest level of instruction possible. It's time to invest in yourself and receive the refresher training you deserve from professionals who understand how to facilitate your advancement.
Whether you're an ethics committee member, clinical research staff member, or a student embarking on a career in clinical research, our training programs are tailored to meet your needs and set you up for success in the dynamic field of clinical research in 2024.
Good Clinical Practice Training Certificate
How can CCRPS propel your career in 2024?
At CCRPS, we're dedicated to equipping individuals in or aspiring to join the clinical trial industry with the necessary knowledge and training conveniently accessible at their fingertips. We understand that committing hours to classroom study isn't always feasible amidst your professional responsibilities. That's why we've developed our comprehensive GCP refresher course, designed to be completed entirely online.
Enrolling in our course offers you the opportunity to delve into advanced-level content to prepare for your Good Clinical Practice Certification testing.
What benefits await you upon enrollment in our practice training?
While GCP certification is mandatory for all clinical research professionals, its significance is particularly pronounced for certain groups:
Investigators representing drug companies, research centers, hospitals, and more.
Members of ethics committees.
Clinical research staff, including clinical research associates, coordinators, trial managers, etc.
Students aspiring to enter the clinical research industry.
Who should consider enrolling in CCRPS's groundbreaking training courses?
Whether you're already a seasoned clinical research professional or aspiring to become one, global recommendations underscore the importance of receiving GCP training and certification. Beyond being a minimum requirement, GCP certification holds several key advantages:
It serves as a formal international validation of an individual's eligibility to work in the clinical research field.
Organizations and companies rely on GCP-certified professionals to ensure compliance with industry regulations and guidelines.
GCP training imparts essential knowledge of clinical research regulations to participants.
Pharmaceutical, biotech companies, and contract research organizations prioritize hiring GCP-certified employees.
In the ever-evolving landscape of the clinical research industry in 2024, ICH GCP training has never been more crucial. Stay ahead of the curve and ensure your knowledge remains current with CCRPS's online practice training courses.
Experience the transformative impact on your clinical research career with our innovative approach to preparation and practice. Elevate your skills and stay abreast of industry developments with CCRPS today!
Download our ICH GCP Attestation Form

Good Clinical Practice GCP Training
Click this link to demo our ICH GCP training free online here!
Good Clinical Practice Training Certificate Syllabus
Introduction to Clinical Research, ICH GCP, and CFR
An Introduction to Clinical Research
An Overview of ICH GCP
Code of Federal Regulations
CFR 21 Part 11
Roles and Responsibilities (Sponsor/CRO, IRB, and Investigator)
Sponsor/CRO Responsibilities
13 Principles, IRB, & Investigator Roles
Informed Consent & Patient Safety
Adverse Event Reporting & Responsibilities
Reporting Responsibilities of the Investigators
Adverse Events
Ethical Research in Vulnerable Populations
Ethics of Research Involving Children
Ethics of Research Involving Pregnant Women and Fetuses
Ethics of Research Involving Prisoners
Trial Management, Data Handling, and Record Retention
Trial Management – Data Handling and Record Retention
Common Terminology Used In Clinical Research
Commonly Used Abbreviations and Terms in Clinical Research
ICH GCP Certification
ICH GCP Certification Exam
ICH GCP Resources
E6(R2) Good Clinical Practice: Integrated Addendum to ICH E6(R1)
FDA resource for E6 r2 addendum (also included in course)
Good Clinical Practice Resource Guide
Division of Microbiology and Infectious Diseases December 2015
WHO Library Cataloguing-in-Publication Data Handbook for good clinical research practice (GCP):
Guidance for implementation
Linkedin Resource of ICH GCP related jobs and roles
Latest trials, website updates, and more
Take courses from CCRPS and learn more on how to become a clinical research professional.
Discover more from Clinical Research Training | Certified Clinical Research Professionals Course
ICH GCP Guidelines
The ICH GCP guidelines provide public assurance that trial subjects' rights, security and well-being are protected in accord with the principles which have their source from Helsinki Declaration. Compliance ensures credible clinical data; 15 points present a unified standard for European Union (EU), Japan & United States to facilitate mutual acceptance by regulatory authorities across those jurisdictions currently compliant with WHO's good practices along side Australia Canada Nordic countries+World Health Organization
This principle has been developed with all their current good clinical practices of the European Union, Japan and USA in addition to those from Australia Canada Nordic countries World Health Organization (WHO). The guidelines established within this document may also be applied during other types medical trials which could have an effect on individual subjects' safety or well being.
ICH GCP Training Free
Here are some ICH GCP training free online guidelines. Make a quizlet or copy the ich gcp guidelines quizlet, then manually rewrite each of these into an individual card that you can easily remember because it's all on one page! To review them more effectively – combine any two cards together if they both pertain to something related in theme (examples: "Committee" & “Implementation”). Continue condensing words and combining sections until down from 50-100 flashcards; after doing so take time out every day this week before your exam(s) for practice reviewing what has been learned thus far by going through each set again slowly but thoroughly while listening carefully
Now you can get internationally accredited ICH GCP certification for $50 through CCRPS course which includes several examples in each video to solidify your knowledge.
ICH GCP Guidelines
The ICH GCP guidelines, including ich gcp e6, provides public assurance that the rights, security and also well-being of trial subjects are protected in accord with the principles which have their source in the Declaration of Helsinki. In addition, compliance ensures credible clinical trial data. The 15 ICH GCP principles presents a unified standard for the European Union (EU), Japan and the United States to facilitate the mutual acceptance of clinical data by the regulatory authorities in those jurisdictions. The principle has been developed with all their current good clinical practices of the European Union, Japan, and the USA, in Addition to those of Australia, Canada, the Nordic countries and the World Health Organization (WHO). This principle ought to be followed when generating. The principles established in this guideline may also be applied to other clinical investigations which might have an influence on the security and well-being of individual subjects.
ICH GCP Training Free
In order to self-learn ich gcp training free online:
1) make a quizlet account (or use the ich gcp guidelines quizlet)
2) manually rewrite each of the guidelines below into quizlet (this is ESSENTIAL in getting the guidelines to stick in your brain!)
3) continue condensing the words and combining guidelines until you’re down to 50-100 flashcards
4) review set 2-3 times and delete cards to clearly remember
5) continue to review and delete cards until you have it memorized!
While our ICH GCP training course Is only $50 it is essential to learning to applying ich gcp guidelines in an advanced method, you should be able to remotely memorize the guidelines on your own for free as an expert adult learner.
ICH GCP GLOSSARY
While our ICH GCP training course is essential to learning to applying ich gcp guidelines in an advanced method, you should be able to remotely memorize the guidelines on your own for free as an expert adult learner.
1. ICH GCP GLOSSARY
1.1 Adverse Drug Reaction (ADR)
From the pre-approval clinical experience with a new medicinal product or its new usages, particularly as the therapeutic dose(s) could not have been established: all noxious and unintended responses to a medicinal product related to any dose ought to be considered adverse drug reactions. The term responses to a medicinal product means that a causal relationship between a medicinal product and an adverse event is at least a reasonable chance, i.e. the connection can't be ruled out. Regarding marketed medicinal products: a reaction to a drug that is noxious and unintended and that occurs at doses normally utilized in man for prophylaxis, diagnosis, or treatment of diseases or for modification of physiological function (see the ICH Guideline for Clinical Safety Data Management: Definitions and Standards for Expedited Reporting).
1.4 Applicable Regulatory Requirement(s)
Any regulation (s) and law (s) addressing the conduct of clinical trials of investigational products.
1.5 Approval (in relation to Institutional Review Boards)
The affirmative decision of the IRB that the clinical trial was reviewed and could be conducted at the institution site within the constraints set forth by the IRB, the institution, Good Clinical Practice (GCP), and the relevant regulatory requirements.
1.7 Audit Certificate
A statement of confirmation by the auditor that an audit has happened. 1.9 Audit Trail
Documentation which allows reconstruction of the class of occasions. 1.10 Blinding/Masking
A process where a couple of parties into this trial are kept unaware of the treatment assignment(s). Single-blinding usually indicates the topic (s) being unaware, and also double-blinding usually indicates the topic (s), investigator(s), track, and, sometimes, data analyst(s) being unaware of the treatment assignment(s).
1.11 Case Report Form (CRF)
A printed, optical, or electronic document designed to record all the protocol required data to be recorded to the sponsor on each trial field.
1.12 Clinical Trial/Study
Any investigation in human subjects meant to discover or verify the clinical, psychiatric or other pharmacodynamic effects of nvestigational product(s), or to identify any adverse reactions to an investigational product(s), or to research absorption, distribution, metabolism, and excretion of an investigational product(s) together with the goal of ascertaining its security and/or effectiveness.
1.13 Clinical Trial/Study Report
A written outline of some trial/study of any therapeutic, prophylactic, or diagnostic agent conducted in human subjects, where the clinical and statistical description, presentations, and analyses are fully integrated into one report (see the ICH Guideline for Structure and Content of Clinical Study Reports).
1.14 Comparator (Product)
An investigational or marketed product (i.e., active control), or placebo, used as a benchmark in a clinical investigation.
1.15 Compliance (in relation to trials)
Adherence to all of the trial-related needs, Good Clinical Practice (GCP) requirements, and the relevant regulatory requirements.
1.17 Deal
A written, dated, and signed agreement between two or more involved parties that sets out any arrangements on delegation and distribution of tasks and duties , if appropriate, on financial issues. The protocol could serve as the foundation of a contract.
1.18 Coordinating Committee
A committee that a sponsor may organize to coordinate with the behavior of a multicentre trial.
1.19 Coordinating Investigator
An employee assigned the responsibility of the coordination of investigators at several centers participating in a multicentre trial.
1.20 Contract Research Organization (CRO)
A individual or a business (commercial, academic, or other) contracted by the sponsor to do at least one of a host's trial-related responsibilities and purposes.
1.21 Immediate Access
Permission to examine, analyze, verify, and reproduce any records and reports which are important to analysis of a medical trial. Any celebration (e.g., national and international regulatory authorities, sponsor's monitors and auditors) with direct access should take all reasonable measures within the constraints of the applicable regulatory requirement(s) to keep the confidentiality of subjects' identities and sponsor's proprietary information.
1.22 Documentation
All documents, in any kind (such as, but not restricted to, written, digital, magnetic, and optical records, and tests, x-rays, and electrocardiograms) that describe or record the methods, behavior, or effects of a trial, and the factors affecting a trial, and the action taken.
1.23 Critical Documents
Documents that individually and collectively permit evaluation of the behavior of a study and the quality of the data generated.
1.24 Good Clinical Practice (GCP)
A benchmark for the design, conduct, performance, monitoring, auditing, recording, analyses, and reporting of clinical trials that offers assurance that the data and reported results are credible and accurate, and the rights, ethics, and confidentiality of trial subjects are protected.
1.25 Independent Data-Monitoring Committee (IDMC/Data and Safety Monitoring Board, Monitoring Committee, Data Monitoring Committee)
A separate data-monitoring committee which could be determined by the sponsor to assess at intervals the progress of a clinical trial, the safety information, and the critical efficacy endpoints, and to recommend to the sponsor whether to continue, change, or discontinue a trial.
1.26 Impartial Witness
A person, who's independent of this trial, that can't be unfairly influenced by people associated in this trial, who attends the informed consent process if the subject or the subject's legally acceptable representative can't read, and who reads the informed consent form and any other written information provided to the topic.
1.27 Independent Ethics Committee (IEC)
An independent body (a review board or a committee, institutional, regional, national, or supranational), constituted of caregivers and non-medical associates, whose duty is to make sure the security of their rights, security and well-being of human issues involved in an investigation and to provide public assurance of the protection, by, among other things, reviewing and approving / providing appropriate view on, the trial procedure, the arrangement of the investigator(s), facilities, and the processes and material to be utilized in obtaining and documenting informed consent of the trial subjects.
1.28 Informed Consent
A procedure in which a subject voluntarily confirms his or her willingness to take part in a specific trial, after being informed of all details of the trial that relate to the subject of choice to engage. Informed consent is documented by way of a written, signed and dated informed consent form.
1.29 Inspection
The action by a regulatory authority(ies) of conducting an official review of documents, records, facilities, and some other sources which are deemed by the authority(ies) to be associated with the clinical trial which could be found in the website of this trial, in the host's or contract study organization's (CRO's) facilities, or at other establishments deemed appropriate by the regulatory authority(ies).
1.30 Institution (medical)
Any private or public entity or agency or medical or dental facility where clinical trials have been conducted.
1.31 Institutional Review Board (IRB)
An independent body constituted of medical, scientific, and non-scientific associates, whose duty is to guarantee the security of their rights, security and well-being of human subjects involved in a trial by, among other things, reviewing, approving, and providing continuing review of trial protocol and amendments and of the methods and substance to be utilized in obtaining and documenting informed consent of the trial subjects.
1.33 Investigational Merchandise
A pharmaceutical form of an active ingredient or placebo being tested or used as a benchmark in a clinical trial, such as a product with a marketing authorization when used or assembled (formulated or packaged) in a sense different from the approved form, or if used for an unapproved indication, or when used to get additional information regarding an approved use.
1.34 Partner
A individual accountable for the behavior of this clinical trial at a trial website. When a trial has been conducted by a group of people at a trial site, the investigator is the responsible leader of the group and might be known as the researcher. See also Subinvestigator.
1.35 Investigator / Institution
An expression meaning "the investigator and/or institution, where required by the applicable regulatory requirements".
1.36 Investigator's Brochure
A compilation of the clinical and nonclinical data on the investigational product(s) which is relevant to the study of the investigational product(s) in human subjects (see 7. Investigator’s Brochure).
1.37 Legally Acceptable Representative
An individual or juridical or other body authorized under applicable law to consent, on behalf of a prospective subject, to the subject's participation in the clinical trial.
1.38 Monitoring
The act of overseeing the progress of a clinical trial, and of ensuring that it is conducted, recorded, and reported in accordance with the protocol, Standard Operating Procedures (SOPs), Good Clinical Practice (GCP), and the applicable regulatory requirement(s).
1.39 Monitoring Report
A written report from the monitor to the sponsor after each site visit and/or other trial-related communication according to the sponsor’s SOPs.
1.40 Multicentre Trial
A clinical trial conducted according to a single protocol but at more than one site, and therefore, carried out by more than one investigator.
1.41 Nonclinical Study
Biomedical studies not performed on human subjects.
1.42 Opinion (in relation to Independent Ethics Committee)
The judgement and/or the advice provided by an Independent Ethics Committee (IEC). 1.43 Original Medical Record
See Source Documents (Below in 1.52).
1.44 Protocol
A document that describes the objective(s), design, methodology, statistical considerations, and organization of a trial. The protocol usually also gives the background and rationale for the trial, but these could be provided in other protocol referenced documents. Throughout the ICH GCP Guideline the term protocol refers to protocol and protocol amendments.
1.45 Protocol Amendment
A written description of a change(s) to or formal clarification of a protocol. 1.46 Quality Assurance (QA)
All those planned and systematic actions that are established to ensure that the trial is performed and the data are generated, documented (recorded), and reported in compliance with Good Clinical Practice (GCP) and the applicable regulatory requirement(s).
1.47 Quality Control (QC)
The operational techniques and activities undertaken within the quality assurance system to verify that the requirements for quality of the trial-related activities have been fulfilled.
1.48 Randomization
The process of assigning trial subjects to treatment or control groups using an element of chance to determine the assignments in order to reduce bias.
1.49 Regulatory Authorities
Bodies with the power to regulate. In the ICH GCP guideline the expression Regulatory Authorities includes the authorities that review submitted clinical data and those that conduct inspections (see 1.29). These bodies are sometimes referred to as competent authorities.
1.50 Serious Adverse Event (SAE) or Serious Adverse Drug Reaction (Serious ADR)
Any untoward medical occurrence that at any dose: - results in death, - is life-threatening, - requires inpatient hospitalization or prolongation of existing hospitalization, - results in persistent or significant disability/incapacity, or - is a congenital anomaly/birth defect (see the ICH Guideline for Clinical Safety Data Management: Definitions and Standards for Expedited Reporting).
1.51 Source Data
All information in original records and certified copies of original records of clinical findings, observations, or other activities in a clinical trial necessary for the reconstruction and evaluation of the trial. Source data are contained in source documents (original records or certified copies).
1.52 Source Documents
Original documents, data, and records (e.g., hospital records, clinical and office charts, laboratory notes, memoranda, subjects' diaries or evaluation checklists, pharmacy dispensing records, recorded data from automated instruments, copies or transcriptions certified after verification as being accurate copies, microfiches, photographic negatives, microfilm or magnetic media, x-rays, subject files, and records kept at the pharmacy, at the laboratories and at medico-technical departments involved in the clinical trial).
1.53 Sponsor
An individual, company, institution, or organization which takes responsibility for the initiation, management, and/or financing of a clinical trial.
1.54 Sponsor-Investigator
An individual who both initiates and conducts, alone or with others, a clinical trial, and under whose immediate direction the investigational product is administered to, dispensed to, or used by a subject. The term does not include any person other than an individual (e.g., it does not include a corporation or an agency). The obligations of a sponsor-investigator include both those of a sponsor and those of an investigator.
1.55 Standard Operating Procedures (SOPs)
Detailed, written instructions to achieve uniformity of the performance of a specific function.
1.56 Subinvestigator Any individual member of the clinical trial team designated and supervised by the investigator at a trial site to perform critical trial-related procedures and/or to make important trial-related decisions (e.g., associates, residents, research fellows). See also Investigator.
1.57 Subject/Trial Subject
An individual who participates in a clinical trial, either as a recipient of the investigational product(s) or as a control.
1.58 Subject Identification Code
A unique identifier assigned by the investigator to each trial subject to protect the subject's identity and used in lieu of the subject's name when the investigator reports adverse events and/or other trial related data.
1.59 Trial Site
The location(s) where trial-related activities are actually conducted. 1.60 Unexpected Adverse Drug Reaction
An adverse reaction, the nature or severity of which is not consistent with the applicable product information (e.g., Investigator's Brochure for an unapproved investigational product or package insert/summary of product characteristics for an approved product) (see the ICH Guideline for Clinical Safety Data Management: Definitions and Standards for Expedited Reporting).
1.61 Vulnerable Subjects
Individuals whose willingness to volunteer in a clinical trial may be unduly influenced by the expectation, whether justified or not, of benefits associated with participation, or of a retaliatory response from senior members of a hierarchy in case of refusal to participate. Examples are members of a group with a hierarchical structure, such as medical, pharmacy, dental, and nursing students, subordinate hospital and laboratory personnel, employees of the pharmaceutical industry, members of the armed forces, and persons kept in detention. Other vulnerable subjects include patients with incurable diseases, persons in nursing homes, unemployed or impoverished persons, patients in emergency situations, ethnic minority groups, homeless persons, nomads, refugees, minors, and those incapable of giving consent.
1.62 Well-being (of the trial subjects)
The physical and mental integrity of the subjects participating in a clinical trial.
2. THE PRINCIPLES OF ICH GCP
2.1 Clinical trials should be conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki, and that are consistent with GCP and the applicable regulatory requirement(s).
2.2 Before a trial is initiated, foreseeable risks and inconveniences should be weighed against the anticipated benefit for the individual trial subject and society. A trial should be initiated and continued only if the anticipated benefits justify the risks.
2.3 The rights, safety, and well-being of the trial subjects are the most important considerations and should prevail over interests of science and society.
2.4 The available nonclinical and clinical information on an investigational product should be adequate to support the proposed clinical trial.
2.5 Clinical trials should be scientifically sound, and described in a clear, detailed protocol.
2.6 A trial should be conducted in compliance with the protocol that has received prior institutional review board (IRB)/independent ethics committee (IEC) approval/favourable opinion.
2.7 The medical care given to, and medical decisions made on behalf of, subjects should always be the responsibility of a qualified physician or, when appropriate, of a qualified dentist.
2.8 Each individual involved in conducting a trial should be qualified by education, training, and experience to perform his or her respective task(s).
2.9 Freely given informed consent should be obtained from every subject prior to clinical trial participation.
2.10 All clinical trial information should be recorded, handled, and stored in a way that allows its accurate reporting, interpretation and verification.
2.11 The confidentiality of records that could identify subjects should be protected, respecting the privacy and confidentiality rules in accordance with the applicable regulatory requirement(s).
2.12 Investigational products should be manufactured, handled, and stored in accordance with applicable good manufacturing practice (GMP). They should be used in accordance with the approved protocol.
2.13 Systems with procedures that assure the quality of every aspect of the trial should be implemented.
3. INSTITUTIONAL REVIEW BOARD/INDEPENDENT ETHICS COMMITTEE (IRB/IEC)
3.1 Responsibilities
3.1.1 An IRB/IEC should safeguard the rights, safety, and well-being of all trial subjects. Special attention should be paid to trials that may include vulnerable subjects.
3.1.2 The IRB/IEC should obtain the following documents: trial protocol(s)/amendment(s), written informed consent form(s) and consent form updates that the investigator proposes for use in the trial, subject recruitment procedures (e.g. advertisements), written information to be provided to subjects, Investigator's Brochure (IB), available safety information, information about payments and compensation available to subjects, the investigator’s current curriculum vitae and/or other documentation evidencing qualifications, and any other documents that the IRB/IEC may need to fulfil its responsibilities. The IRB/IEC should review a proposed clinical trial within a reasonable time and document its views in writing, clearly identifying the trial, the documents reviewed and the dates for the following: - approval/favourable opinion; - modifications required prior to its approval/favourable opinion; - disapproval / negative opinion; and - termination/suspension of any prior approval/favourable opinion.
3.1.3 The IRB/IEC should consider the qualifications of the investigator for the proposed trial, as documented by a current curriculum vitae and/or by any other relevant documentation the IRB/IEC requests.
3.1.4 The IRB/IEC should conduct continuing review of each ongoing trial at intervals appropriate to the degree of risk to human subjects, but at least once per year. 3.1.5 The IRB/IEC may request more information than is outlined in paragraph 4.8.10 be given to subjects when, in the judgement of the IRB/IEC, the additional information would add meaningfully to the protection of the rights, safety and/or well-being of the subjects.
3.1.6 When a non-therapeutic trial is to be carried out with the consent of the subject’s legally acceptable representative (see 4.8.12, 4.8.14), the IRB/IEC should determine that the proposed protocol and/or other document(s) adequately addresses relevant ethical concerns and meets applicable regulatory requirements for such trials.
3.1.7 Where the protocol indicates that prior consent of the trial subject or the subject’s legally acceptable representative is not possible (see 4.8.15), the IRB/IEC should determine that the proposed protocol and/or other document(s) adequately addresses relevant ethical concerns and meets applicable regulatory requirements for such trials (i.e. in emergency situations).
3.1.8 The IRB/IEC should review both the amount and method of payment to subjects to assure that neither presents problems of coercion or undue influence on the trial subjects. Payments to a subject should be prorated and not wholly contingent on completion of the trial by the subject.
3.1.9 The IRB/IEC should ensure that information regarding payment to subjects, including the methods, amounts, and schedule of payment to trial subjects, is set forth in the written informed consent form and any other written information to be provided to subjects. The way payment will be prorated should be specified.
3.2 Composition, Functions and Operations
3.2.1 The IRB/IEC should consist of a reasonable number of members, who collectively have the qualifications and experience to review and evaluate the science, medical aspects, and ethics of the proposed trial. It is recommended that the IRB/IEC should include: (a) At least five members. (b) At least one member whose primary area of interest is in a nonscientific area. (c) At least one member who is independent of the institution/trial site. Only those IRB/IEC members who are independent of the investigator and the sponsor of the trial should vote/provide opinion on a trial-related matter. A list of IRB/IEC members and their qualifications should be maintained.
3.2.2 The IRB/IEC should perform its functions according to written operating procedures, should maintain written records of its activities and minutes of its meetings, and should comply with GCP and with the applicable regulatory requirement(s).
3.2.3 An IRB/IEC should make its decisions at announced meetings at which at least a quorum, as stipulated in its written operating procedures, is present.
3.2.4 Only members who participate in the IRB/IEC review and discussion should vote/provide their opinion and/or advise.
3.2.5 The investigator may provide information on any aspect of the trial, but should not participate in the deliberations of the IRB/IEC or in the vote/opinion of the IRB/IEC.
3.2.6 An IRB/IEC may invite nonmembers with expertise in special areas for assistance.
3.3 Procedures
The IRB/IEC should establish, document in writing, and follow its procedures, which should include:
3.3.1 Determining its composition (names and qualifications of the members) and the authority under which it is established.
3.3.2 Scheduling, notifying its members of, and conducting its meetings. 3.3.3 Conducting initial and continuing review of trials.
3.3.4 Determining the frequency of continuing review, as appropriate.
3.3.5 Providing, according to the applicable regulatory requirements, expedited review and approval/favourable opinion of minor change(s) in ongoing trials that have the approval/favourable opinion of the IRB/IEC.
3.3.6 Specifying that no subject should be admitted to a trial before the IRB/IEC issues its written approval/favourable opinion of the trial.
3.3.7 Specifying that no deviations from, or changes of, the protocol should be initiated without prior written IRB/IEC approval/favourable opinion of an appropriate amendment, except when necessary to eliminate immediate hazards to the subjects or when the change(s) involves only logistical or administrative aspects of the trial (e.g., change of monitor(s), telephone number(s)) (see 4.5.2).
3.3.8 Specifying that the investigator should promptly report to the IRB/IEC: (a) Deviations from, or changes of, the protocol to eliminate immediate hazards to the trial subjects (see 3.3.7, 4.5.2, 4.5.4). (b) Changes increasing the risk to subjects and/or affecting significantly the conduct of the trial (see 4.10.2). (c) All adverse drug reactions (ADRs) that are both serious and unexpected. (d) New information that may affect adversely the safety of the subjects or the conduct of the trial.
3.3.9 Ensuring that the IRB/IEC promptly notify in writing the investigator/institution concerning: (a) Its trial-related decisions/opinions. (b) The reasons for its decisions/opinions. (c) Procedures for appeal
ICH GCP
GCP Online Course: Advanced ICH GCP
Certification (AGCPC)
ENROLL
What Is ICH GCP Certification
Home Course Catalog
Career Guides
Group Orders
Enroll
of its decisions/opinions.
of its decisions/opinions.
3.4 Records
The IRB/IEC should retain all relevant records (e.g., written procedures, membership lists, lists of occupations/affiliations of members, submitted documents, minutes of meetings, and correspondence) for a period of at least 3 years after completion of the trial and make them available upon request from the regulatory authority(ies). The IRB/IEC may be asked by investigators, sponsors or regulatory authorities to provide its written procedures and membership lists.
4. INVESTIGATOR
4.1 Investigator's Qualifications and Agreements
4.1.1 The investigator(s) should be qualified by education, training, and experience to assume responsibility for the proper conduct of the trial, should meet all the qualifications specified by the applicable regulatory requirement(s), and should provide evidence of such qualifications through up-to-date curriculum vitae and/or other relevant documentation requested by the sponsor, the IRB/IEC, and/or the regulatory authority(ies).
4.1.2 The investigator should be thoroughly familiar with the appropriate use of the investigational product(s), as described in the protocol, in the current Investigator's Brochure, in the product information and in other information sources provided by the sponsor.
4.1.3 The investigator should be aware of, and should comply with, GCP and the applicable regulatory requirements.
4.1.4 The investigator/institution should permit monitoring and auditing by the sponsor, and inspection by the appropriate regulatory authority(ies).
4.1.5 The investigator should maintain a list of appropriately qualified persons to whom the investigator has delegated significant trial-related duties.
4.2 Adequate Resources
4.2.1 The investigator should be able to demonstrate (e.g., based on retrospective data) a potential for recruiting the required number of suitable subjects within the agreed recruitment period.
4.2.2 The investigator should have sufficient time to properly conduct and complete the trial within the agreed trial period.
4.2.3 The investigator should have available an adequate number of qualified staff and adequate facilities for the foreseen duration of the trial to conduct the trial properly and safely.
4.2.4 The investigator should ensure that all persons assisting with the trial are adequately informed about the protocol, the investigational product(s), and their trial-related duties and functions.
4.3 Medical Care of Trial Subjects
4.3.1 A qualified physician (or dentist, when appropriate), who is an investigator or a sub-investigator for the trial, should be responsible for all trial-related medical (or dental) decisions.
4.3.2 During and following a subject's participation in a trial, the investigator/institution should ensure that adequate medical care is provided to a subject for any adverse events, including clinically significant laboratory values, related to the trial. The investigator/institution should inform a subject when medical care is needed for intercurrent illness(es) of which the investigator becomes aware.
4.3.3 It is recommended that the investigator inform the subject's primary physician about the subject's participation in the trial if the subject has a primary physician and if the subject agrees to the primary physician being informed.
4.3.4 Although a subject is not obliged to give his/her reason(s) for withdrawing prematurely from a trial, the investigator should make a reasonable effort to ascertain the reason(s), while fully respecting the subject's rights.
4.4 Communication with IRB/IEC
4.4.1 Before initiating a trial, the investigator/institution should have written and dated approval/favourable opinion from the IRB/IEC for the trial protocol, written informed consent form, consent form updates, subject recruitment procedures (e.g., advertisements), and any other written information to be provided to subjects.
4.4.2 As part of the investigator's/institution’s written application to the IRB/IEC, the investigator/institution should provide the IRB/IEC with a current copy of the Investigator's Brochure. If the Investigator's Brochure is updated during the trial, the investigator/institution should supply a copy of the updated Investigator’s Brochure to the IRB/IEC.
4.4.3 During the trial the investigator/institution should provide to the IRB/IEC all documents subject to review.
4.5 Compliance with Protocol
4.5.1 The investigator/institution should conduct the trial in compliance with the protocol agreed to by the sponsor and, if required, by the regulatory authority(ies) and which was given approval/favourable opinion by the IRB/IEC.
The investigator/institution and the sponsor must sign the protocol, or another contract, to confirm arrangement.
4.5.2 The investigator shouldn't implement any deviation from, or modifications of this protocol without agreement by the sponsor and prior review and documented approval/favourable opinion in the IRB/IEC of an amendment, except where necessary to eliminate an immediate hazard(s) to trial subjects, or when the change(s) involves only logistical or administrative aspects of the trial (e.g., alter in track (s), change of phone number(s)).
4.5.4 The investigator may implement a deviation from, or a reversal of, the protocol to eliminate an immediate hazard(s) to trial subjects without previous IRB/IEC approval/favourable opinion. When possible, the implemented deviation or change, the causes of this, also, if appropriate, the proposed protocol amendment(s) ought to be filed: (a) into the IRB/IEC for inspection and approval/favourable view, (b) to the sponsor for agreement and, when necessary, (c) into the regulatory authority(ies).
4.6.2 Where allowed/required, the investigator/institution may/should assign some or all the investigator's/institution's duties for investigational product(s) accountability at the trial site(s ) ) to an proper pharmacist or another suitable person who's under the oversight of their investigator/institution. .
4.6.3 The investigator/institution or a pharmacist or other appropriate person, who's given by the investigator/institution, should keep records of their item's delivery to the trial site, the inventory at the website, the usage by each topic, and also the yield to the sponsor or alternative disposition of unused product(s). These records must include dates, numbers, batch/serial numbers, expiration dates (if applicable), and the exceptional code numbers assigned to the investigational product(s) and trial subjects. Researchers should keep records that document adequately that the subjects were provided the doses specified by the protocol and reconcile all investigational product(s) obtained from the host.
4.6.4 The investigational product(s) ought to be kept as defined by the host (see 5.13.2 and 5.14.3) and in compliance with applicable regulatory requirement(s).
4.6.5 The investigator should ensure that the investigational product(s) are utilized only in compliance with the accepted protocol.
4.6.6 The investigator, or a person designated by the investigator/institution, must describe the proper use of the investigational product(s) to each subject and should check, at times appropriate for the trial, that each subject is following the directions correctly. If the trial is blinded, the investigator should promptly document and explain to the sponsor any early unblinding (e.g., accidental unblinding, unblinding because of a serious adverse event) of the investigational product(s).
4.8 Informed Consent of Trial Subjects
4.8.1 In obtaining and documenting informed consent, the investigator must comply with the applicable regulatory requirement(s), and should adhere to GCP and to the ethical principles which have their source in the Declaration of Helsinki. Before the start of the trial, the investigator needs to have the IRB/IEC's composed approval/favourable view of this written informed consent form and any other written information to be offered to subjects.
4.8.2 The written informed consent form and any other written information to be given to subjects must be revised whenever important new information becomes available that may be pertinent to this subject's approval. Any revised written informed consent form, and written advice must get the IRB/IEC's approval/favourable view ahead of usage. The subject or the subject's legally acceptable representative ought to be informed in a timely fashion if new information becomes available that may be pertinent to the subject's willingness to continue participation in the trial. The communication of the information ought to be documented.
4.8.5 The investigator, or a person designated by the investigator, should fully inform the subject or, even if the topic is not able to give informed consent, the subject's legally acceptable representative, of all pertinent aspects of the trial including the written advice and also the approval/ favourable opinion by the IRB/IEC.
4.8.6 The language used in the written and oral information regarding the trial, including the written informed consent form, must be non-technical as functional and should be clear to the subject or the subject's legally acceptable representative and the impartial witness, where applicable.
4.8.7 Before informed consent may be obtained, the investigator, or someone designated by the investigator, should offer the subject or the subject's legally acceptable representative ample time and opportunity to ask about details of the trial and also to choose whether or not to take part in the trial. All queries concerning the trial ought to be answered to the satisfaction of the topic or the subject's legally acceptable representative.
4.8.8 Before a subject's involvement in the analysis, the written informed consent form ought to be signed and dated by the topic or from the subject's legally appropriate agent, and from the man who conducted the informed consent discussion.
4.8.9 If a topic can't read or if a legally acceptable representative is not able to read, an impartial witness should be present throughout the entire informed consent discussion. Following the written informed consent form and any other written information to be given to subjects, will be read and explained to the subject or the subject's legally acceptable representative, and after the subject or the subject's legally acceptable representative has orally consented to the subject of involvement in the trial and, if capable of doing this, has signed and dated the informed consent form, the witness must sign and personally date the consent form. (b) The intention of the trial. If there's not any intended clinical benefit to the subject, the topic ought to be made aware of the (I) The alternative procedure(s) or course(s) of treatment which could be accessible to the matter, and their important potential benefits and hazards. (j) The reimbursement and/or therapy readily available to the subject at case of trial-related injury. (l) The anticipated expenses, if any, to the subject of participating in the trial. (n) The monitor(s), the auditor(s), the IRB/IEC, along with the regulatory authority(ies) will be granted direct entry to the subject's original medical records for verification of clinical trial processes and/or information, without violating the confidentiality of this topic, to the extent allowed by the applicable legislation and regulations and that, by signing a written informed consent form, the subject or the subject's legally acceptable representative is authorizing such access. (o) That records identifying the subject will be kept confidential and, to the extent allowed by regulations or laws, won't be made publicly accessible. If the outcomes of the trial have been published, the subject's identity will stay confidential. (p) The subject or the subject's legally acceptable representative will be informed in a timely manner if information becomes available that may be pertinent to the subject's willingness to continue participation in the trial. (q) The individual (s) to contact for more information concerning the trial and the rights of trial subjects, and whom to contact in case of trial-related injury. (r) The foreseeable conditions and/or motives under the subject's involvement in the trial could be terminated. (s) The expected duration of the subject's involvement in the trial. (t) The approximate number of subjects included with the trial.
4.8.11 Prior to participation in the trial, the subject or the subject's legally acceptable representative should be given a copy of the signed and dated written informed consent form and any other written information supplied to the topics. During a subject's involvement in the trial, the subject or the subject's legally acceptable representative should get a copy of the signed and dated consent form updates and a copy of any amendments to the written information given to subjects.
4.8.12 When a clinical trial (therapeutic or non-therapeutic) includes subjects who can only be registered in the trial with the permission of the subject's legally acceptable representative (e.g., minors, or individuals with severe dementia), the subject ought to be informed about the trial to the extent compatible with the subject of comprehension and, if capable, the subject should sign and personally date the written informed consent.
4.8.13 Except as described in 4.8.14, a non-therapeutic trial (i.e. a trial where there is not any anticipated direct clinical benefit to the subject), needs to be conducted in subjects who give consent and that sign and date the written informed consent form.
4.8.14 Non-therapeutic trials might be conducted in subjects with consent of a legally acceptable representative provided that the following requirements are fulfilled: (a) The aims of the trial can't be fulfilled by way of a trial in subjects that can provide informed consent. (c) The adverse influence on the subject's well-being is reduced and minimized. (d) The trial isn't prohibited by legislation. (e) The approval/favorable view of this IRB/IEC is especially sought on the inclusion of these topics, and the written approval/ favorable opinion covers this aspect. Topics in such trials must be particularly closely monitored and must be removed if they seem to be overly distressed.
4.8.15 In crisis situations, when prior permission of the subject isn't feasible, the permission of the subject's legally acceptable representative, if present, should be asked. When previous consent of the topic isn't feasible, along with the subject's legally acceptable representative isn't available, enrollment of this topic ought to require steps described in the protocol or elsewhere, with recorded approval/favorable opinion from the IRB/IEC, to safeguard the rights, security and well-being of this topic and also to guarantee compliance with applicable regulatory requirements. The subject or the subject's legally acceptable representative ought to be informed about the trial when possible and agree to continue along with additional approval as appropriate (see 4.8.10) ought to be asked.
4.9 Records and Reports
4.9.1 The investigator should ensure the accuracy, completeness, legibility, and timeliness of their information reported to the host at the CRFs and in all necessary reports.
4.9.2 Data reported on the CRF, which are derived from source documents, ought to be in accordance with the source documents or the discrepancies should be clarified.
4.9.3 Any alteration or correction to a CRF ought to be dated, initialed, and explained (if necessary) and shouldn't obscure the original entry (i.e. an audit trail ought to be preserved ); that applies to both written and electronic changes or corrections (visit 5.18.4 (n)). Sponsors should provide advice to investigators or the researchers' designated representatives on making such corrections. Sponsors must have written procedures to ensure corrections or changes in CRFs created by sponsor's designated agents are recorded, are needed, and are backed by the investigator. The investigator must maintain records of the corrections and changes.
4.9.4 The investigator/institution must keep the trial documents as stated in Essential Documents for the Conduct of a Clinical Trial (see 8.) The investigator/institution must take steps to avoid accidental or premature destruction of those records.
4.9.5 Essential documents should be retained until at least two years following the final approval of a marketing application in an ICH region and until there are no pending or contemplated marketing applications in an ICH region or at least two years have elapsed since the formal discontinuation of clinical development of the investigational item. These records should be kept for a longer period however if required by the relevant regulatory requirements or by having an arrangement with the host. It's the obligation of the host to notify the investigator/institution concerning when these documents no longer have to be kept (see 5.5.12).
4.9.6 The financial details of the trial ought to be recorded in an agreement between the host and the investigator/institution.
4.9.7 Upon request of the monitor, auditor, IRB/IEC, or regulatory authority, the investigator/institution must make readily available for direct access all requested trial-related documents.
4.10 Progress Reports
4.10.1 The investigator must submit written summaries of this trial status to the IRB/IEC yearly, or more often, if asked by the IRB/IEC.
4.10.2 The investigator should promptly provide written reports on the host, the IRB/IEC (see 3.3.8) and, where applicable, the institution on any changes significantly affecting the behavior of this trial, or raising the risk to subjects. The follow-up and immediate reports must identify subjects by unique code numbers assigned to the trial subjects instead of from the subjects' names, personal identification numbers, or addresses. The investigator must also comply with the applicable regulatory requirement(s) associated with the reporting of unexpected serious adverse drug reactions to the regulatory authority(ies) along with the IRB/IEC.
4.11.2 Adverse laboratory or events abnormalities identified in the protocol as critical to safety evaluations should be reported to the host in line with the coverage requirements and within the time intervals specified by the host in the protocol.
4.12 Premature Termination or Suspension of a Trial
In case the trial is prematurely terminated or suspended for any reason, the investigator/institution should immediately inform the trial issues, should guarantee proper treatment and follow-up for those issues, and, where required by the applicable regulatory requirement(s), should notify the regulatory authority(ies). Additionally:
4.12.1 If the investigator terminates or suspends a trial without prior agreement of the host, the investigator must inform the institution where applicable, and also the investigator/institution should immediately notify the host and the IRB/IEC, and should offer the sponsor along with the IRB/IEC a detailed written explanation of the termination or suspension.
4.12.2 If the sponsor terminates or suspends a trial (see 5.21), the investigator must immediately notify the institution where applicable along with the investigator/institution should immediately inform the IRB/IEC and supply the IRB/IEC a detailed written explanation of the termination or suspension.
4.12.3 When the IRB/IEC terminates or suspends its approval/favorable view of a trial (see 3.1.2 and 3.3.9), the investigator must inform the institution where applicable along with the investigator/institution should immediately notify the host and supply the sponsor with a detailed written explanation of the termination or suspension.
4.13 Final Report(s) by Investigator
Upon completion of the trial, the investigator, where relevant, should notify the institution; the investigator/institution must offer the IRB/IEC using a review of the trial's result, and the regulatory authority(ies) with any reports required.
5. SPONSOR
5.1 Quality Assurance and Quality Control
5.1.1 The host is responsible for implementing and maintaining quality assurance and quality management systems with written SOPs to make certain that trials are conducted and data are generated, documented (recorded), and documented according to the protocol, GCP, and the applicable regulatory requirement(s).
5.1.2 The host is responsible for securing agreement from all involved parties to ensure immediate access (see 1.21) to each of trial related websites, origin data/documents, and reports for the purpose of monitoring and auditing by the sponsor, and review by domestic and international regulatory authorities.
5.1.3 Quality control ought to be applied to every point of data handling to make sure that all data are reliable and have been processed properly.
5.1.4 Agreements, created by the host with all the investigator/institution and some other parties involved with the clinical trial, must be in writing, within this protocol or in another arrangement.
5.2 Contract Research Organization (CRO)
5.2.1 A sponsor may transfer any or all the host's trial-related responsibilities and functions to a CRO, but the ultimate responsibility for its integrity and quality of the trial data always resides with the host. The CRO should apply quality assurance and quality management.
5.2.2 Any trial-related responsibility and function that's transferred to and assumed by a CRO ought to be given in writing.
5.2.3 Any trial-related responsibilities and functions not specifically transferred to and assumed by a CRO are retained by the host.
5.2.4 All references to a host within this guideline apply to a CRO to the extent that a CRO has assumed the trial related duties and functions of a host.
5.3 Medical Experience
The sponsor must designate suitably qualified medical staff that are easily available to counsel trial related health questions or issues. If needed, external advisor (s) can be made for this function.
5.4 Trial Design
5.4.1 The host must use qualified people (e.g. biostatisticians, clinical pharmacologists, and physicians) as appropriate, during all phases of their trial process, in designing the protocol and CRFs and preparing the investigations into assessing and preparing interim and final clinical trial reports.
5.5 Trial Management, Data Handling, and Record Keeping i.e. ICH GCP guidelines for clinical data management
5.5.1 The host must use appropriately qualified people to supervise the general conduct of this trial, to deal with the information, to confirm the information, to conduct the statistical analyses, and also to prepare the demo reports. The IDMC should have written operating procedures and keep written records of its meetings.
5.5.3 When using electronic trial data management and/or remote electronic trial information programs, the host needs to: (a) Ensure and document that the electronic data processing procedure(s) adheres to the sponsor's established requirements for completeness, accuracy and reliability, and consistent intended performance (i.e. identification ). (b) Maintains SOPs for utilizing such systems. (c) Make sure that these systems are intended to allow data changes in such a manner in which the data changes are documented and that there isn't any deletion of input data (i.e. keep an audit trail, information path, edit path ). (d) Keep a safety system which prevents unauthorized access into this information. (e) Keep a listing of those people that are licensed to produce information modifications (see 4.1.5 and 4.9.3). (g) Shield the blinding, if some (e.g. keep the data during data entry and processing system ).
5.5.4 When data are transformed during processing, then it must remain possible to evaluate the initial observations and data with the data that is processed.
5.5.5 The host must utilize an unambiguous subject identification code (visit 1.58) which enables identification of all of the information reported for every topic.
5.5.6 The host, along with other owners of all this information, should keep each the sponsor-specific necessary documents of interest to the trial (see 8).
5.5.7 The sponsor must maintain all sponsor-specific necessary files in conformance with all the applicable regulatory requirement(s) of this country(ies) in which the item is accepted, or at which the sponsor intends to submit an application for approval(s).
5.5.9 If the sponsor discontinues the clinical development of an investigational solution, the sponsor must notify all of the trial investigators/institutions and most of the regulatory authorities.
5.5.10 Any transfer of possession of this information must be reported on the proper authority(ies), according to the applicable regulatory requirement(s).
5.5.12 The sponsor must notify the investigator(s)/association(s) in writing of their requirement for document retention and should inform the investigator(s)/association(s) in writing whenever the trial associated documents are no more needed.
5.6 Investigator Choice
5.6.1 The host is responsible for picking the investigator(s)/association (s). Each investigator ought to be qualified by experience and training and should have sufficient funds (see 4.1, 4.2) to properly conduct the trial where the investigator is chosen. If business of some coordinating committee or choice of coordinating investigator(s) will be used in multicentre trials, their company and/or choice are the host's responsibility.
5.6.2 Before entering an agreement with an investigator/institution to perform a trial, the sponsor must offer the investigator(s)/association (s) using the routine and also an up-to-date Investigator's Brochure, and should provide adequate time for your investigator/institution to assess the protocol and also the info supplied.
5.6.3 The sponsor must obtain the investigator's/institution's arrangement: (a) to conduct the trial according to GCP, together with all the applicable regulatory requirement(s) (see 4.1.3), also with the protocol agreed to by the host and given approval/favorable remark by the IRB/IEC (see 4.5.1); (b) to comply with processes for information recording/reporting; (c) to allow tracking, auditing and review (see 4.1.4) and (d) to keep the trial associated essential files until the host informs the investigator/institution that these records are no longer required (see 4.9.4 along with also 5.5.12). The host and the investigator/institution need to sign the protocol, or an alternate file, to verify this arrangement.
5.7 Allocation of Duties
Before initiating a trial, the sponsor should define, establish, and devote most of of trial-related responsibilities and purposes.
5.8 Compensation to Subjects and Investigators
5.8.1 If required by the applicable regulatory requirement(s), the host must offer insurance or if indemnify (valid and fiscal policy ) that the investigator/the association against claims arising out of the trial, and except for claims that arise from prosecution and/or neglect.
5.8.2 The host's policies and procedures must deal with the expenses of treatment for trial issues in case of trial-related accidents in agreement with the applicable regulatory requirement(s).
5.8.3 When identification subjects receive reimbursement, the procedure and way of reimbursement must comply with applicable regulatory requirement(s).
5.9 Funding
The financial facets of the trial ought to be recorded in an agreement between the host and the investigator/institution.
5.10 Notification/Submission into Regulatory Authority(ies)
Prior to initiating the clinical investigation (s), the host (or the host and the investigator, even when necessary by the applicable regulatory requirement(s)) must submit any necessary program (s) into the proper authority(ies) for inspection, approval, and/or consent (as needed by the applicable regulatory requirement(s)) to commence the trial(s). Any notification/submission ought to be dated and include adequate information to recognize the routine. (b) A statement obtained in the IRB/IEC it is organized and functions in accordance with GCP and the applicable regulations and laws. (c) Documented IRB/IEC approval/favourable view and, when requested by the host, a recent backup of protocol, written informed consent form(s) and any other written information to be offered to areas, subject recruiting processes, and records associated with payments and reimbursement available to the topics, and some other files the IRB/IEC could have asked.
5.11.2 If the IRB/IEC states its approval/favourable view upon modification (s) in almost any feature of the trial, including alteration (s) of this protocol, written informed consent form and any other written information to be offered to areas, or other processes, the sponsor must obtain in the investigator/institution that a duplicate of the modification(s) created and the date approval/favourable remark was given from the IRB/IEC.
5.11.3 The sponsor must obtain in the investigator/institution dates and documentation of any IRB/IEC reapprovals/re-evaluations with hierarchical view, also of any withdrawals or suspensions of all approval/favourable view.
5.12 Information on Investigational Product(s) 5.12.1 When planning trials, the sponsor must ensure that adequate safety and efficacy information from nonclinical clinical or studies trials are readily available to support human vulnerability from the path, in the doses, for its length, and at the trial population to be analyzed.
5.12.2 The host must upgrade the Investigator's Brochure as important new information becomes available (see 7).
5.13 Manufacturing, Packaging, Labeling, and Coding Investigational Product(s)
5.13.1 The host should ensure that the investigational product(s) (such as active comparator(s) and placebo( if appropriate ) is distinguished as appropriate for the stage of growth of the item (s), is fabricated according to any relevant GMP, and can be coded and tagged in a way that safeguards the blinding, if appropriate. Additionally, the labelling must comply with all applicable regulatory requirement(s).
5.13.2 The sponsor must determine, for the investigational product(s), decent storage temperatures, storage requirements (e.g. protection against mild ), storage times, reconstitution fluids and processes, and apparatus for product extract, if any. The host should notify all parties that are involved (e.g. tracks, researchers, pharmacistsand storage managers) of those determinations.
5.13.3 The investigational product(s) ought to be packed to avoid contamination and improper deterioration during storage and transport.
5.13.4 In clinical trials, the programming system to its investigational product(s) must incorporate a mechanism which allows rapid identification of their item (s) if a health crisis, but doesn't permit imperceptible fractures of this blinding.
5.13.5 If significant formulation changes are produced in the investigational or comparator product(s) throughout the course of clinical improvement, the outcomes of some further studies of the formulated product(s) (e.g. stability, dissolution rate, bioavailability) required to evaluate whether these changes could significantly alter the pharmacokinetic profile of this item ought to be available before using this newest formula in clinical trials.
5.14 Supplying and Handling Investigational Product(s)
5.14.1 The host is responsible for providing the investigator(s)/association (s) using all the investigational product(s ) ).
5.14.2 The host shouldn't provide an investigator/institution using the investigational product(s) before the host obtains all necessary documentation (e.g. approval/favorable view from IRB/IEC and regulatory authority(ies)).
5.14.3 The host must ensure that written procedures contain directions the investigator/institution must follow to the storage and handling of investigational product(s) for your trial and documentation . The processes should address decent and safe receipt, handling, storage, unloading, recovery of fresh product in issues, and yield of unused investigational product(s) to the host (or other disposition if approved by the host and in accordance with all the applicable regulatory requirement(s)).
5.14.4 The host needs to: (a) guarantee timely delivery of investigational product(s) into this investigator(s). (b) Keep records that document dispatch, receipt, disposition, reunite, and also destruction of this investigational product(s) (see 8). (c) Maintain a method for regaining investigational products and recording that this recovery (e.g. for deficient product remember, recover after trial completion( expired merchandise recover ).
5.14.5 The host needs to: (a) Take action to make certain that the investigational product(s) are steady over the length of usage. (b) Maintain adequate quantities of the investigational product(s) utilized from the trials to reconfirm specifications, so should it be necessary, and keep records of batch sample investigations and attributes. To the degree equilibrium allows, samples must be kept either before the investigations of the trial data will be complete or as needed by the applicable regulatory requirement(s), whichever reflects the longer retention interval.
5.15 Record Access
5.15.1 The host must ensure it is given in the protocol or other written agreement which the investigator(s)/association (s) offer immediate access to source data/documents such as trial- related observation, Tests, IRB/IEC inspection, and regulatory review.
5.15.2 The host must verify that every subject has agreed, in writing, to guide accessibility to their own first medical records to get trial-related observation, audit, IRB/IEC inspection, and regulatory scrutiny.
5.16.2 The sponsor must immediately notify all concerned investigator(s)/association (s) and the regulatory authority(ies) of findings which could affect negatively the security of topics, affect the behavior of this trial, or change the IRB/IEC's approval/favourable view to keep the test.
5.17.3 The sponsor must submit to the regulatory authority(ies) all security upgrades and periodic reports, and according to applicable regulatory requirement(s).
5.18 Tracking
5.18.1 Purpose
The functions of trial monitoring are to confirm: (a) The rights and also well-being of individual subjects are protected. (c) The conduct of the offense will be in accordance with the approved protocol/amendment(s), with GCP, along with all the applicable regulatory requirement(s). (b ) Monitors must be suitably trained, and ought to possess the clinical or scientific knowledge required to track the trial satisfactorily. A track's qualifications must be recorded.
5.18.3 Extent and Nature of Monitoring
The host should ensure that the trials have been adequately tracked. The sponsor must determine the right scope and nature of observation. The conclusion of the scope and nature of monitoring should be determined by factors like the purpose, function, style, complexity, blinding, size, and endpoints of this trial. Generally speaking there's a demand for onsite observation, before, during, and after the trialhowever in extraordinary circumstances the host may decide that central observation in combination with processes such as researchers' meetings and training, and comprehensive written advice can assure proper conduct of the trial in agreement with GCP. Statistically controlled sampling could be an acceptable way of selecting the information to be checked.
5.18.4 Monitor's Responsibilities
The track (s) in compliance with the host's requirements need to make sure that the trial will be conducted and recorded properly by executing the following actions when relevant and essential to this trial and the crime website: (a) Acting as the major field of communication between the host and the investigator. (b) Verifying that the investigator has sufficient qualifications and tools (see 4.1, 4.2, 5.6) and stay adequate throughout the trial period, which facilities, such as labs and equipment, and employees, are sufficient to safely and properly conduct the trial and stay adequate throughout the trial period. (ii) The investigational product(s) are provided exclusively to subjects that are qualified for it and in the protocol given dose(s). (iii) That matters are supplied with necessary education on correctly using, managing, storing, and returning to the investigational product(s). (iv) The reception, use, and yield of this investigational product(s) in the trial sites are regulated and recorded adequately. (v) The disposition of unused investigational product(s) in the trial sites complies with all applicable regulatory requirement(s) and can be in accord with the sponsor. (e) Verifying that written informed consent was obtained prior to each subject's involvement in this trial. (f) Ensuring that the investigator gets the current Investigator's brochure, all records, and all of the trial provides required to conduct the trial properly and also to comply with the applicable regulatory requirement(s). (h) Verifying that the investigator and the investigator's trial staff are currently still doing the given trial purposes, in accord with the protocol along with another written agreement between the host and the investigator/institution, also haven't assigned the functions to unauthorized people. (I) Verifying that the investigator will be enroling only qualified subjects. (j) Reporting the matter recruitment rate. (k) Verifying that source files and other trial documents are true, complete, retained up-to-date and preserved. (Id) Verifying that the investigator provides all of the essential documents, notifications, applications, and admissions, and these records are accurate, comprehensive, timely, legible, dated, and also establish that the trial. (m) Assessing the accuracy and completeness of the CRF entries, source files and other trial-related documents contrary to each other. The monitor especially should confirm that: (I) The information required by the protocol have been reported right about the CRFs and therefore are in accordance with the source files. (ii) Any dose or treatment alterations are well documented for all the trial issues. (iii) Adverse events, concomitant medications and inter-current disorders are reported with regard to the routine in the CRFs. (iv) Visits the subjects don't create, tests which aren't conducted, and tests which aren't performed are reported as such on the CRFs. (n) Informing the inheritance of any CRF entrance mistake, omission, or illegibility. The monitor must ensure that proper adjustments, additions, or deletions are made, obsolete, clarified (if needed ), and initialed by the investigator or from a part of the investigator's trial staff who's licensed to first CRF modifications to your investigator. This consent ought to be documented. (o) Deciding whether adverse events (AEs) are reported over the time intervals required by GCP, the protocol, the IRB/IEC, the host, as well as the applicable regulatory requirement(s). (de) Deciding if the investigator is keeping the vital files (see 8. )
5.18.5 Monitoring Procedures
The track (s) must occur after the host's established written SOPs in addition to those processes which are given by the host for tracking a particular trial.
5.18.6 Monitoring Report
(a) The screen must submit an official report to the host after every trial-site see or trial-related communication. (b) Reports must include the date, website, title of the track, and title of the investigator or other person (s) contacted. (c) Reports must include a summary of the track reviewed along with the track's statements regarding the substantial findings/facts, deviations and deficiencies, decisions and actions taken or to be obtained and/or activities recommended to procure compliance. (d) The follow-up and review of this observation report with all the host ought to be recorded by the host designated agent.
5.19 Audit
When Patrons Execute audits, as a Part of Executing quality assurance, they Ought to Think about: 5.19.1 Purpose
The purpose of a host's audit, that will Be independent of and different from regular monitoring or quality control purposes, is to assess trial behavior and compliance with the protocol, SOPs, GCP, and the applicable regulatory requirements.
5.19.2 Selection and Qualification of Auditors
(a) The sponsor must appoint individuals, that are independent of their clinical trials/systems, to run research. (b) The sponsor should make sure that the auditors are qualified by experience and training to conduct audits properly. An auditor's qualifications must be recorded.
5.19.3 Auditing Procedures
(a) The sponsor should ensure that the auditing of clinical trials/systems is conducted with respect to the sponsor's written procedures about which to audit, the way to study, the frequency of analysis, as well as the shape and content of reports. (b) The host's audit program and processes for a trial should be directed by the value of the trial to admissions to regulatory authorities, the amount of subjects from the trial, and the form and complexity of the trial, and the amount of threats to the trial issues, along with also any identified issue (s). (c) The findings and observations of the auditor(s) ought to be documented. (d) To maintain the freedom and importance of the audit function, the regulatory authority(ies) shouldn't routinely ask the audit accounts. Regulatory authority(ies) could find entry to an audit report on a case by case basis if signs of critical GCP non-compliance is present, or even in the course of legal proceeding. (e) If required by applicable law or regulation, the host must offer an audit certification.
5.20 Noncompliance
5.20.1 Noncompliance with all the protocol, SOPs, GCP, or relevant regulatory requirement(s) with an investigator/institution, or from member(s)) of their host's staff should result in prompt action from the host to secure compliance.
5.20.2 in the event the observation and/or auditing describes long-term or serious noncompliance on the part of an investigator/institution, then the host must terminate the employee's /institution's involvement at the trial. Once an investigator's/institution's participation is terminated due to noncompliance, the host must notify immediately the regulatory authority(ies).
5.21 Premature Termination or Suspension of a Trial
When a trial is prematurely terminated or suspended, then the host should immediately inform the investigators/institutions, along with the regulatory authority(ies) of their conclusion or suspension and the reason(s) for the termination or suspension. The IRB/IEC also needs to be advised promptly and given the rationale (s) for the termination or suspension from the host or from the investigator/institution, according to the applicable regulatory requirement(s).
5.22 Clinical Trial/Study Reports When the trial has been completed or terminated, the sponsor should make certain that the clinical trial accounts are prepared and supplied to the regulatory agency(ies) according to the applicable regulatory requirement(s). The host must also make sure that the clinical trial reports in advertising programs meet the criteria of this ICH Guideline for Structure and Content of Clinical Study Reports. (NOTE: The ICH Guideline for Structure and Content of Clinical Study Reports reveal that abbreviated study reports may be appropriate in certain instances.)
5.23 Multicentre Trials For multicentre trials, the sponsor must make sure that:
5.23.1 All researchers conduct this trial from strict compliance with the protocol agreed to by the host and, if necessary, from the regulatory authority(ies), also awarded approval/favorable remark by the IRB/IEC.
5.23.2 The CRFs are made to capture the essential information at all multicentre trial websites. For those researchers that are collecting further information, supplemental CRFs must also be supplied that are intended to capture the extra data.
5.23.3 The duties of coordinating investigator(s) along with another participating investigators are recorded before the beginning of the trial.
5.23.4 All researchers are given directions on after the protocol, to complying with a uniform set of criteria for the evaluation of clinical and laboratory findings, and on finishing the CRFs.
6. But site specific advice might be given on separate protocol page(s), or handled in another agreement, and a few of the info listed below can be included in other protocol referenced documents, including an Investigator's Brochure. Any modification (s) must also bear the amendment number(s) and date(s).
6.1.3 Name and name of the individual (s) authorized to sign the protocol and the protocol change (s) for your host.
6.1.4 Name, name, address, and phone number(s) of their host's medical practitioner (or dentist when appropriate) for the offense.
6.1.5 Name and name of the investigator(s)) who is/are responsible for conducting the trial, along with the address and phone number(s) of the trial site(s).
6.1.6 Name, name, address, and phone number(s)) of the qualified physician (if appropriate), who's accountable for many trial-site associated medical (or dental) decisions (if other than investigator).
6.1.7 Title (s) and address(es) of the clinical laboratory(ies) and other technical or medical section (s) and/or associations involved with the trial.
6.2 Background Information
6.2.1 Title and description of the investigational product(s).
6.2.2 A list of findings in nonclinical studies that potentially have clinical significance and from clinical trials which are linked to this trial.
6.2.3 Summary of the known and possible risks and advantages, if any, to human subjects.
6.2.5 An announcement that the trial will be run in accordance with the protocol, GCP and the applicable regulatory requirement(s).
6.2.6 Description of the population to be researched.
6.2.7 References to literature and information which are related to the trial, which provide background for your trial.
6.3 Trial Objectives and Purpose
A comprehensive outline of the goals and the objectives of the trial.
6.4 Trial Design
The scientific integrity of this trial and the trustworthiness of the information from the trial depend considerably on the trial layout. A description of the trial design, must contain:
6.4.1 A particular statement of the principal endpoints and the secondary endpoints, if any, to be measured throughout the trial.
6.4.2 An outline of this type/design of trial must be performed (e.g. double sided, placebo-controlled( parallel design) and a schematic diagram of trial design, processes and phases.
6.4.3 A description of the measures required to minimize/avoid prejudice, such as: (a) Randomization.
6.4.4 An outline of the trial treatment(s) and the dose and dose regimen of the investigational product(s).
6.4.5 The expected duration of subject participation, and a description of this order and duration of all trial periods, such as followup, if any.
6.4.6 A description of the"stopping rules" or"discontinuation criteria" for different topics, elements of trial and complete trial.
6.4.9 The identification of any data to be recorded directly on the CRFs (i.e. no previous written or electronic record of data), also also to be regarded as source data. (b) The type and timing of this information to be collected for withdrawn subjects. (d) The followup to subjects withdrawn from investigational product treatment/trial therapy.
6.6 Treatment of Topics
6.6.1 The remedy (s) has to be treated, including the name(s) of the item (s), the dose(s), the dosing schedule(s), the route/mode(s) of government, and the treatment period(s), for instance, follow-up interval (s) for subjects for each investigational product treatment/trial therapy group/arm of this trial.
6.6.2 Medicine (s)/treatment(s) permitted (including rescue medication) and not allowed before or throughout the trial.
6.7.2 Methods and timing for assessing, recording, and assessing of efficiency parameters. 6.8.2 The timing and methods for assessing, recording, and assessing safety parameters. 6.8.4 The kind and length of the followup of subjects after adverse events.
6.9 Statistics
6.9.1 A number of the statistical techniques to be employed, including timing of any planned interim analysis(ses).
6.9.2 the amount of subjects planned to be registered.
6.9.3 the degree of importance to be utilized. 6.9.4 Criteria for the conclusion of this trial.
6.9.6 Procedures for reporting any deviation(s) from the original statistical plan (any form (s) from the original statistical plan ought to be clarified and justified from protocol or in the last report( as appropriate).
6.9.7 The choice of topics must be included in the investigations (e.g. all distinct subjects, all dosed subjects, all eligible subjects, evaluable subjects).
6.10 Direct Access to Source Data/Documents The host must ensure it is given in the protocol or other written agreement that the investigator(s)/association (s) will allow trial-related tracking, audits,
IRB/IEC inspection, and regulatory review (s), providing immediate access to supply data/documents.
IRB/IEC inspection, and regulatory review (s), providing immediate access to supply data/documents. 6.13 Data Handling and Record Keeping
6.14 Financing and Insurance Coverage Financing and insurance if not addressed in a separate arrangement.
6.15 Publication Policy Publication policy, if not handled in another agreement. 6.16 Supplements (NOTE: As the protocol along with the clinical trial/study document are closely linked, additional relevant information is found at the ICH Guideline for Structure and Content of Clinical Study Reports.)
INVESTIGATOR'S BROCHURE
7.1 Introduction
The Investigator's Brochure (IB) is a set of the clinical and nonclinical data on the investigational product(s) which relate to the analysis of the merchandise (s) in human subjects. Its objective is to deliver the researchers and others involved with the trial using all the data to facilitate their comprehension of the rationale behind, and their compliance with, several important features of this protocol, like the dosage, dosage frequency/interval, techniques of management: and security monitoring processes. The IB also gives insight to help the clinical direction of their research subjects throughout the course of this clinical trial. The info ought to be displayed in a concise, simple, objective, balanced, and also non-promotional type that allows an individual clinician, or possible investigator, to comprehend it and create his unbiased risk-benefit evaluation of the appropriateness of the planned trial. Because of this a medically qualified individual should normally take part in the screening of an IB, however, the contents of the IB must be accepted by the areas that created the described information. This guideline delineates the minimal information which needs to be contained within an IB and gives tips for its design. It's anticipated that the kind and degree of information available will change with the period of growth of the investigational item. If the investigational product is promoted and its pharmacology is broadly known by medical professionals, a comprehensive IB might not be vital. Where permitted by law enforcement, a fundamental product information booklet, package leaflet, or data could possibly be an proper choice, given that it comprises comprehensive, current, and comprehensive advice on all parts of the investigational product which may be of significance to this investigator. If a promoted product has been analyzed for a new usage (i.e., a brand new sign ), an IB unique to this new use ought to be ready. The IB must be evaluated at least annually and revised as required in accordance with a host's written procedures. More regular revision could be appropriate based on the phase of evolution and the creation of relevant new info. Nonetheless, in accordance with Good Clinical Nutrition, pertinent new information might be so significant that it needs to be conveyed to the researchers, and maybe into the Institutional Review Boards (IRBs)/Independent Ethics Committees (IECs) or regulatory governments before it's contained in a revised IB. Usually, the host is responsible for ensuring an up- to-date IB is made accessible for the investigator(s) and the researchers are responsible for supplying the up-to-date IB into the accountable IRBs/IECs. In the instance of a worker sponsored trial, then the sponsor-investigator must find out if a booklet is available in the industrial maker. If the investigational product is supplied by this sponsor-investigator, then they must offer the essential info to the trial staff. In scenarios when planning of a proper IB is impractical, the sponsor-investigator must supply, as a replacement, an enlarged background information element in the trial procedure which includes the minimal present data described within this principle.
7.2 General Considerations the IB should comprise:
7.2.1 Title Page
This ought to offer the host's name, the identification of every investigational product (i.e., study number, compound or accepted generic title, and transaction name(s) where legally permissible and desired by the host), along with also the launch date. It's also suggested an edition number, and a reference to this date and number of the variation that it supersedes, be supplied. A good instance is provided in Appendix 1.
7.2.2 Confidentiality Statement
The sponsor might desire to incorporate a statement instructing the investigator/recipients to take care of the IB as a private record for the only information and usage of this investigator's team along with the IRB/IEC.
7.3 Contents of the Investigator's Brochure
The IB should include these segments, each with literature references where appropriate: 7.3.1 Table of Contents An illustration of the Table of Contents is provided at Appendix 2
7.3.2 Summary
A short summary (preferably not exceeding two pages) ought to be granted, highlighting the substantial physical, chemicaland pharmaceutical, pharmacological, toxicological, pharmacokinetic, metabolic, and clinical data available that's pertinent to this point of clinical development of the investigational item.
7.3.3 Introduction
A short introductory statement ought to be provided that includes the compound name (and generic and trade name(s) when accepted ) of the investigational product(s), all active components, the investigational product (s) pharmacological category and its expected location in this category (e.g. benefits ), the justification for performing study using an investigational product(s), as well as the expected prophylactic, therapeutic, or diagnostic sign (s). At length, the introductory statement must offer the overall strategy to be followed in assessing the investigational item.
7.3.4 Physical, Chemical, and Pharmaceutical Properties and Formulation
A description ought to be given of this investigational product material (s) (such as the compound or structural formulation (e), plus a succinct summary ought to be due to the applicable physical, chemical, and pharmaceutical properties. To allow proper security measures to be obtained in the duration of this trial, an outline of the formula (s) to be utilized, such as excipients, ought to be supplied and justified when clinically applicable. Directions to the storage and management of this dose form(s) must also be granted. Any similarities with other substances should be noted.
ICH GCP E6 R2
On Mar 8, 2018, the FDA updated ICH E6(R1) with E6(R2) Good Clinical Practice: Integrated Addendum
to ICH E6(R1). Here are some noticeable changes and how they will impact the industry.
1. One of the key improvement is the new definition of a licensed copy of a situation report form (1.11). This improved definition states:"[a] newspaper or digital copy of the first document that's been confirmed (e.g., with a dated signature) or was generated via a validated procedure to make an specific copy using all the very exact features and data as the first." This improvement helps better explain how to ascertain the validity of trial-related documents duplicates, such as source files.
Additionally, the definition of tracking (1.38) has been broadened to incorporate the observation program, which can be described as "[an] outline of these methods, duties, and demands for tracking the trial." The updated definition doesn't call for the monitoring plan for a standalone file, but leaves an expectation that a proper plan is different. In addition, the observation report (1.39) definition has expanded to add "[results] of almost virtually any centralized monitoring also needs to be noted." Nonetheless many patrons now include concentrated monitoring as part of the general monitoring procedures because if this monitoring doesn't happen through a formal onsite observation trip, it might not be satisfactorily documented. The expanded definition will guarantee that patrons produce an account to demonstrate the concentrated monitoring that has been performed.
A number of improvements have been suggested to the Investigator department (part 4). for one, part 4.2.6 has been updated to say:"[in] the event the investigator/institution keeps the assistance of any party to do research jobs, they ought to ensure this celebration is qualified to execute these research jobs and should employ procedures to guarantee the integrity of their analysis jobs completed and any information created." These qualifications and oversight responsibilities weren't explicitly mentioned in the preceding edition, however, clinical trial sponsors anticipated researchers to implicitly stick to those guidelines. The upgraded statements today represent FDA's well- established advice on the pupil's supervisory responsibilities.
The definition of sudden adverse drug response (1.60) currently contains a brand new definition titled "identification of automatic systems" (1.60.1). But, there doesn't appear to be an evident connection between the definition of adverse medication reactions and this definition--"[a] practice of establishing and recording the specified prerequisites of a computerized system may be always fulfilled. An logical step will be to create this new PC validation definition 1.61 then renumber the past two definitions from the Glossary (vulnerable themes and well-being) into 1.62 and 1.63, respectively, and which could possibly be performed when the last record is published.
Part 5.18.6 (Tracking Report) contains a new section (e) that says "[tracking] results should be supplied to the host (such as proper management and personnel accountable for trial and website supervision) in a timely fashion for review and follow up as indicated. Outcomes of monitoring activities must be recorded in enough detail to permit confirmation of compliance with the observation program."
Section 5, Sponsors, comprises substantial adjustments and enhancements. The draft comprised a significant new segment 5.0 (Quality Management), where the notions of quality management, with a focus on risk management, are incorporated into the host's responsibilities. Though risk management procedures are well-known in the healthcare care sector, they have yet to be extensively applied to the preparation and execution of clinical trials. The upgrade will call for clinical trials patrons to start obtaining the essential instruction and tools to establish those principles. Two helpful overall resources include ICH Q9, Quality Risk Management, that will be a high level summary of risk management fundamentals, along with ISO 14971, Application of Risk Management to Medical Devices, a worldwide security standard related to all phases in the life span of a medical apparatus, for example its early growth. While these two records are tailored toward producing hazard management, they can provide useful information related to clinical trial preparation too. Table 1 lists the entire text of this suggested quality control department.
The draft creates no suggested modifications to the Department 3, Institutional Overview Board/Independent Ethics Committee.
In section 4.9, Records and Reports, a fresh introductory announcement (4.9.0) was added which says "[the] investigator must keep accurate and adequate source records and trial documents which have all applicable observations on each of the website's trial topics. Source data ought to be conducive, legible, contemporaneous, first, authentic, and complete. Changes to supply data ought to be traceable, shouldn't obscure the original entrance, and ought to be clarified if required (e.g., through an audit trail)."
Regular review of data that is submitted. Identification of lost information, conflicting data, information outliers, or sudden deficiency of variability and protocol deviations which could possibly be indicative of significant or systematic mistakes in data collection and reporting in a website or through sites, or might be indicative of possible data manipulation or data integrity issues. Utilization of statistical investigations to identify information trends like the consistency and range of information within and across websites. Evaluation of website features and performance metrics. Choice of websites and/or procedures are targeted onsite monitoring.
The previous modification increases section 8.1 (Introduction) the following improvements:"[the] host and investigator/institution need to keep a listing of the place(s) of the individual key documents.’ The storage method (no matter the media used) need to supply for record identification, research, and recovery. Based upon the actions being completed, individual trials will call for additional files not particularly mentioned in the vital document listing. The host or investigator/institution should incorporate these within this trial master document. The host should make sure that the investigator has command of continuous access to the CRF information reported to the host. The host shouldn't have management of these data. When a backup is utilized to replace a first record, the backup should meet the prerequisites for certified copies.
The draft includes several alterations that address fluctuations from the scale, sophistication, and expense of clinical trials because the former version was embraced. Since clinical research workers have access to innovative technologies and risk management procedures that might raise efficacy and concentrate on important clinical research actions, E6 has been amended to promote the implementation of advanced and more effective methods to clinical trial design, conduct, supervision, documenting, and reporting, while still ensuring human subject security and information integrity are preserved. Additionally, the upgrade includes changes to describe criteria on electronic documents and documents that are essential. In the end, the new record is intended to assist clinical research protect human areas, keep data integrity and quality, and correctly record trial benefits. This guide will emphasize the vital changes that impact research professionals. These alterations are anticipated to be assessed and approved inside ICH and then integrated into the E6 record from the end of 2016.
Revisions to the segment on tracking (5.18) reflect a stronger dependence on risk-based observation. The revisions include the components in the FDA's recent advice on risk-based observation. These alterations have been noted in part 5.18.3 (Extent and Nature of Monitoring) and comprise these improvements:"The host must create a systematic, guaranteed, risk-based method of tracking clinical trials. The flexibility at the scope and character of monitoring described in this section is meant to enable diverse approaches that enhance the efficacy and efficiency of observation. A combo of onsite and concentrated monitoring actions could be proper. The sponsor must file the rationale behind the selected observation approach (e.g., from the monitoring program )."
Part 5.20 (Noncompliance) comes with an augmentation which reflects regulatory ability expectations which patrons will try to recognize the root of non-compliance at a strong way and execute effective corrective and preventative strategies. The new segment (5.20.1) says:"[when] important noncompliance is found, the host must execute a root cause investigation and execute appropriate corrective and preventative actions. When required by law or regulation, then the host must notify the regulatory authority(ies) whenever the noncompliance is a severe violation of the trial procedure or GCP."
The draft also suggested a new segment 5.18.7 (Monitoring Plan) that says:"The host must develop a monitoring program that's tailored to the particular human subject security and information integrity risks of this trial. The program must describe the monitoring approach, the monitoring responsibilities of all of the parties involved, the a variety of tracking methods to be utilized, and the justification for their usage. The program should also highlight the observation of essential data and procedures. Particular care ought to be given to all these aspects which aren't regular clinical practice which need further training. The monitoring program should reference the related policies and processes."
Section 5.2, Contract Research Organization (CRO)also comprises two suggested changes that need sponsors to have a more active part in tackling their CROs. Section 5.2.1 was improved with the following announcement:"[the] host must ensure oversight of almost any trial-related responsibilities and works performed on its own behalf." Section 5.2.2 was improved with the following announcement:"[the] host must record approval of some subcontracting of all trial-related responsibilities and works with a CRO." This is very related to small and startup manufacturers which rely heavily upon CROs for handling all or most trial-related pursuits. The modifications state that patrons might not abdicate this duty and have to have a more active part in their supervision of the CROs.
Additionally, the ICH Upgrades underline the usage of centralized tracking as a vital approach to match and lower the frequency or extent of onsite observation.
Section 5.5.3 (b) is modified to describe expectations for normal operating procedures (SOPs) for digital data systems and handling. The proposed language says"[the] SOPs must pay for system installation, setup, and usage. The SOPs must explain system identification and performance testing, information collection and handling, program maintenance and system safety measures, shift management, information backup, recovery, contingency planning, and decommissioning. The obligations of the sponsor, employee, as well as other parties connected to the usage of those unmanned systems ought to be apparent, and the consumers must be supplied with instruction in the usage of their systems." The draft also comes with a brand new announcement 5.5.3 (h), which says that patrons are predicted to"[ensure] that the integrity of their information containing any information that explain the context, content, and arrangement of their information." This inclusion is very important whenever making modifications to the automatic systems, including software upgrades or migration of information.
Clinical Research Associate Salary - What's the pay for a clinic research associate?
Here's What You Need to Know to Get a Clinical Research Associate Job
What's the pay for a Clinical Research Associate?: $61-$110K
What's the pay for a Clinical Research Associate?: $61-$110K
A Clinical Research Associate (or Monitor) is hired either in-house (“the trial site”) or externally (by the sponsor or CRO) to review clinical trial data and ensure that investigational therapies are tested ethically and scientifically through performing site visits that review files like patient medical notes in order to ensure quality of trial data. The catch 22 of clinical research associate jobs is that the ICH GCP guidelines require both education AND experience in order to work in this role, so getting your foot in the door is tough. Once you get experience, your education (i.e., certifications or degrees showing understanding of additional responsibilities) can help promotes you quickly through the CRA career ladder. For those looking to understand more about these guidelines, the ICH-GCP course might be a crucial step.
How to become or get promoted as a clinical research associate?
Having a certification through CCRPS’s accredited Advanced Clinical Research Associate Certification (CRA) course can help professionals 1) get promoted 2) get a raise 3) improve efficiency 4) get hired as a Clinical Research Associate. Additionally, those interested in broader roles in clinical research may consider the Clinical Research Coordinator course, the Pharmacovigilance Certification, or the Clinical Trials Assistant Training.
How much does a Clinical Research Associate make in the United States?
The average Clinical Research Associate salary in the United States is $61-110K. Salary ranges can vary widely depending on many important factors, including education, certifications, additional skills, and the number of years you have spent in your profession. For those looking at a leadership role, the Advanced Clinical Research Project Manager Certification or the Advanced Principal Investigator Physician Certification could be relevant. Furthermore, the Medical Monitor Certification can enhance skills for those specifically interested in medical monitoring aspects.
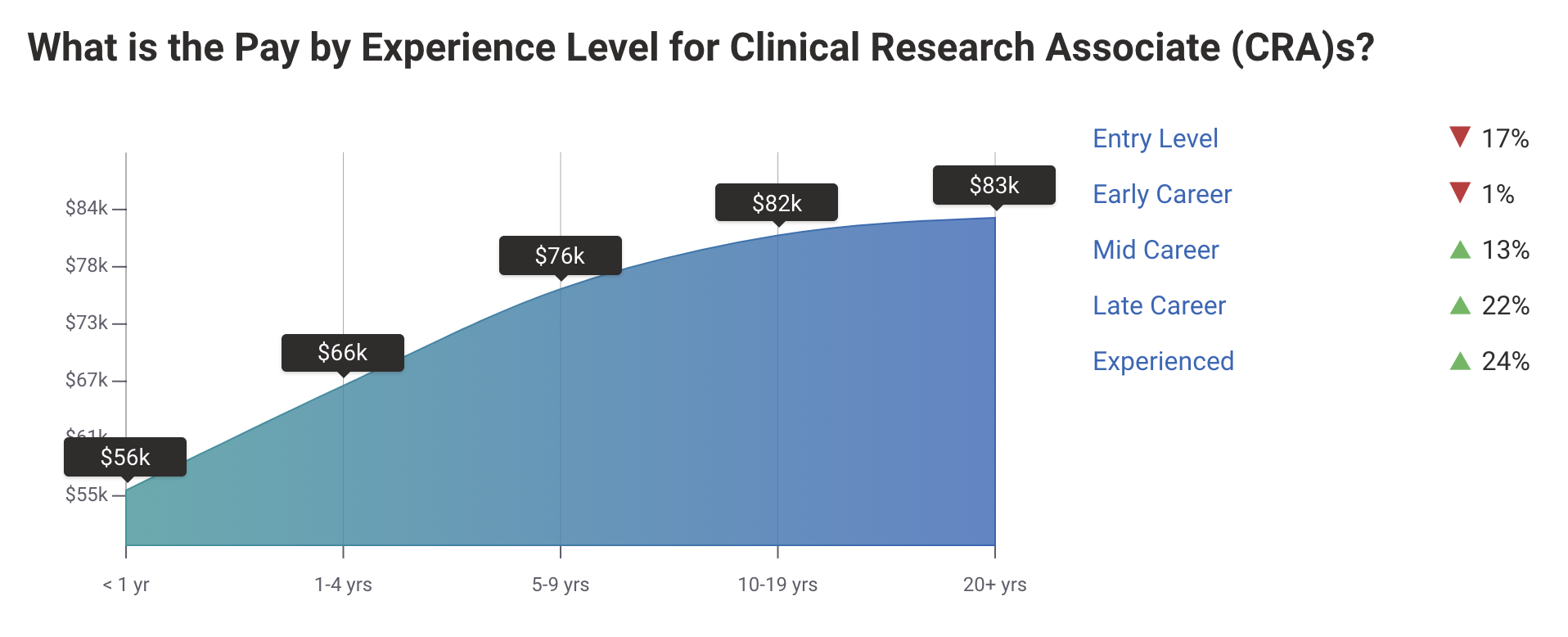
Clinical Research Associate (CRA) Salary
Per Payscale “An entry-level Clinical Research Associate (CRA) with less than 1 year experience can expect to earn an average total compensation (includes tips, bonus, and overtime pay) of $55,588 based on 203 salaries. An early career Clinical Research Associate (CRA) with 1-4 years of experience earns an average total compensation of $66,245 based on 1,118 salaries. A mid-career Clinical Research Associate (CRA) with 5-9 years of experience earns an average total compensation of $76,086 based on 294 salaries. An experienced Clinical Research Associate (CRA) with 10-19 years of experience earns an average total compensation of $81,540 based on 157 salaries. In their late career (20 years and higher), employees earn an average total compensation of $83,342.”
Determine CRA Salary by location using Payscale
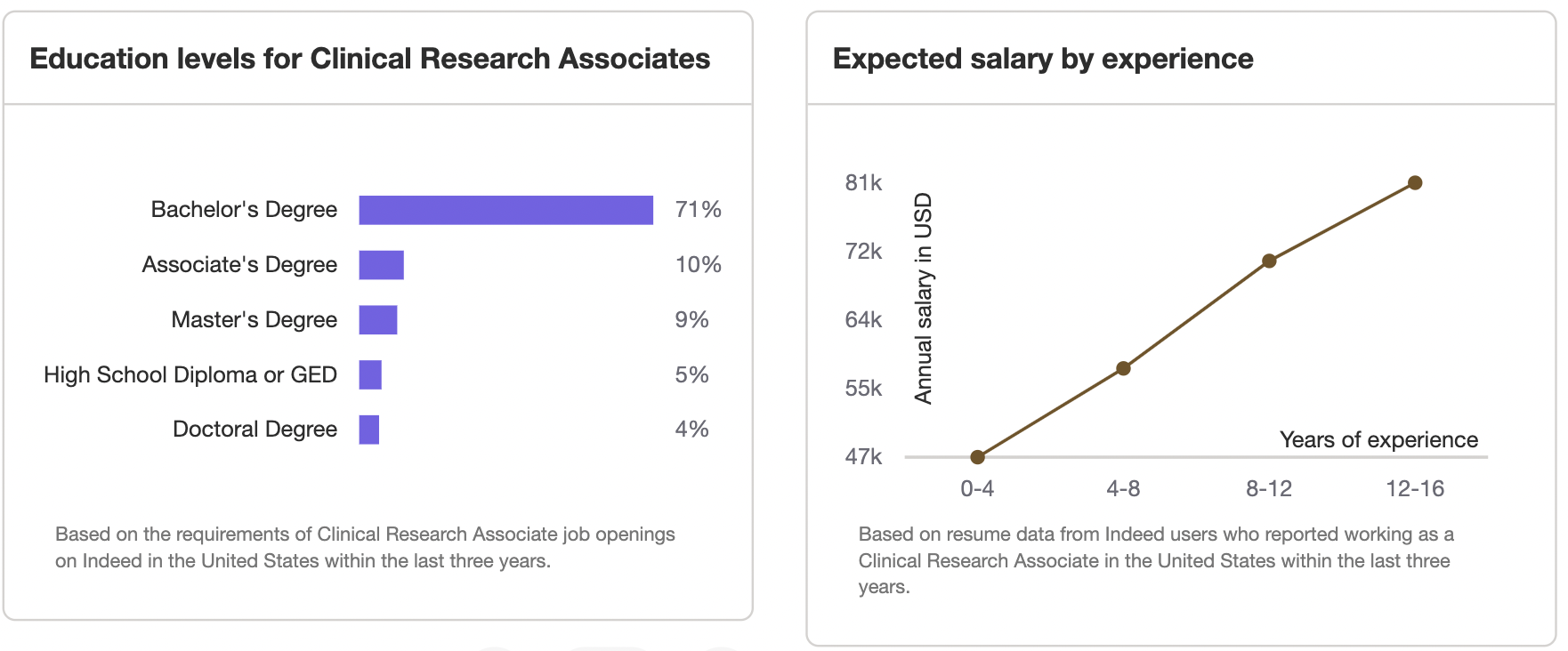
CRA Career Progression - Indeed
Salary: Clinical Research Associate
Research Associate Salary resource: Click here to see the CRA Salary Range from 1,800+ employers
Clinical Research Associates Pay: Clinical Research Associate Salary in United States (by city)
Durham, NC - $97K
New York, NY, Irvine, CA, Houston, TX - $95K
Philadelphia, PA - $92K
Atlanta, GA - $84K
Raleigh, NC - $82K
Chicago, IL - $79K
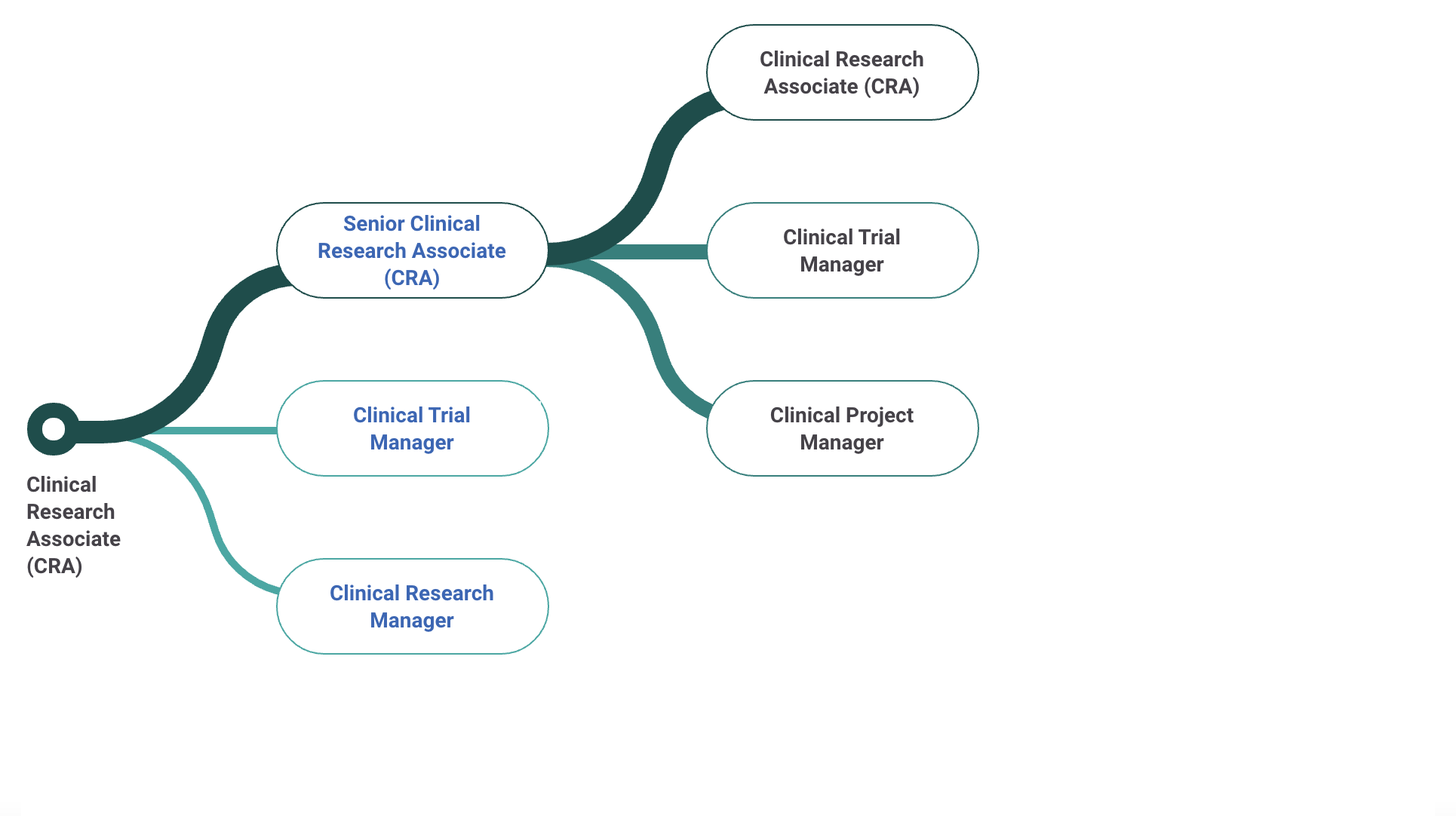
Payscale: CRA Career Pathway
Senior Clinical Research Associate Salary
The average salary of a Senior Senior Clinical Research Associate is ~105K per year ($54/hour, $2k/week, $9k/month). This can range from $81k to $139k.
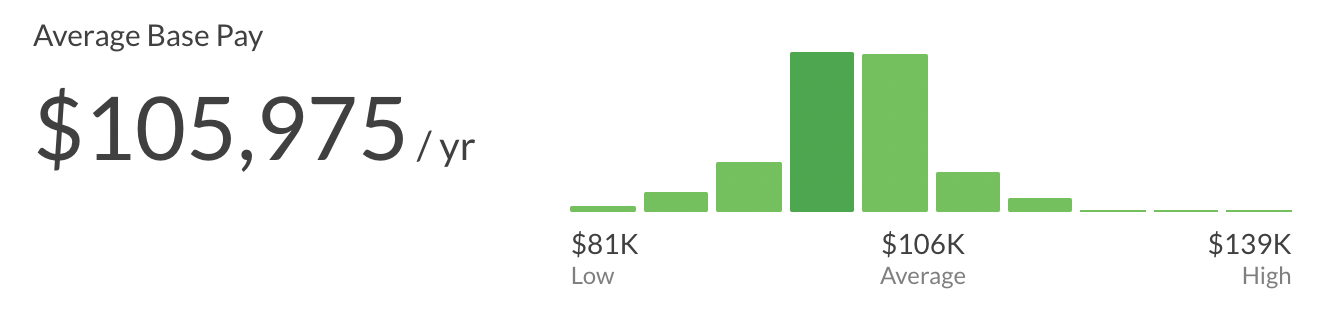
Glassdoor Salary range for Senior CRAs
Starting Salary of a Clinical Research Associate Position is between $60,000 and $65,000
Some employers may prefer hiring entry-level clinical research associates with less experience in clinical research so they can learn their job functions by training under Senior CRAs or CRA IIs.
Clinical Research Associate II Salary is $86,677 / yr
CRA II @ PPD: $84,733/yr
CRA II @ PRA Health Sciences: $96,017/yr
CRA II @ IQVIA: $79,412/yr
CRA II @ ICON: $100,000/yr
Comment below how much you get paid and what helped you get promoted!
Having a certification through CCRPS’s accredited Advanced Clinical Research Associate Certification course can help professionals 1) get promoted 2) get a raise 3) improve efficiency 4) get hired as a Clinical Research Associate.

7 Reasons Why You Should Start a Career in Clinical Research
begin a Career in Clinical Research
How you ever thought that there’s a perfect job out there for you, but you just haven’t found it yet? If you are motivated, informed, and interested in a science and medical career, you might have just found your answer. Many clinical research professionals say this is the golden ticket to a great career in the science field.
Why Clinical Research as a Career
The clinical research industry is a highly lucrative and expanding field. The global clinical trials market has been estimated at $46.8 billion in 2019.
As the push for new vaccines and therapeutics continues to get stronger, the field is expected to grow even more in value. Experts predict that the global market will hit $69.9 billion by 2027. The future in clinical research is bright, and it is one that you will want to be a part of.
Contrary to popular belief, working in clinical research doesn’t have to mean you have to stay in a lab. There are demands and opportunities for every skill set, if you know how to find them. Below, I have put together an in-depth guide on why you should get into clinical research.
Working in Clinical Research
You like a job that’s flexible
Don’t like working in a cubical? How about heading to the airport every morning instead? If you like a job that keeps you moving, then becoming a Clinical Research Associate (CRA) and working in clinical trials might be the right move for you. Learn more about becoming a CRA with this CRA Certification Course.
CRAs, contrary to what most people believe, don’t collect data or interact with patients. A CRA’s day-to-day job is to travel between different research sites and verify data transcription (i.e., data management). CRAs can also become part of the project management as a clinical trial manager of an entire trial.
They are also called “monitors” and a part of "regulatory affairs", because it is also their job and clinical experience to ensure that every site is following proper compliance and protocols.
There are two types of CRAs: home base or in-house. Home base CRAs work remotely. That means they work and travel from home. Most CRAs work for contract research organizations who are hired by sponsors to conduct their multi-site trials.
If you get tired of working home base, you can become an in-house CRA. In-house CRAs stay in one site and work together with a home base CRA to keep each other updated with what is happening at their site.
You like working with people
Have you ever been told that you are a people person with great communication skills? If talking to someone about how you can change their health for the better sounds like something you’d enjoy, you should definitely look into becoming a Clinical Research Coordinator (CRC). Explore our Clinical Research Coordinator Course to get started.
CRCs are the backbones to every project. They conduct patient visits, input source documents into the electronic data capture (EDC), and ensure that every trial is following compliance. They are also responsible for handling regulatory documents and updating the Principal Investigator (PI) with trial results.
CRCs conduct a variety of tasks, all of which impact the progress and development of the trial. Every successful clinical trials team needs is a good CRC. So, if you have strong interpersonal skills and know how to stay organized, you will be an indispensable part of the team.
You are detail-oriented and tech-savvy
Are you a self-proclaimed techie? Perhaps you’ve dabbled in coding, pick up computer programs easily, and maybe even have a background in IT. Technology is the future. If you think you have a knack for organizing data, you should look into becoming a Clinical Trial Assistant (CTA). Check out our Clinical Trials Assistant Training to learn more.
CTAs, also known as Clinical Research Assistants, manage the Trial Master File (TMF). They file, archive, and maintain trial documents and study files. They are also responsible for closing inquiries from the CRA, as well as providing administrative support to the research team. Every important step in clinical research, pre-clinical research, study startup, site management, needs a dependable CTA.
While most jobs in clinical research require some understanding of technology, it is especially important for the CTA to know what they are doing when it comes to managing trial documents and study files. In addition, it is equally important that the CTA is organized and knows how to pay attention to detail.
Working in Clinical Trials
Means you like a good salary...with room for promotion
Though there are many career paths within clinical research, most people begin their career as CTAs or CRCs in entry-level positions. Depending on your skill set and what kind of experiences you can bring to the table, either position will help you get your feet through the door.
According to salary.com, a CTA’s average salary in 2020 is $63,000. They generally earn between a range of $54,300 and $73,000, and are provided with benefits such as healthcare and social security.
If being more hands-on in the research process appeals to you, you might be a good fit for a CRC. Similarly, CRCs are making an average of $63,117 in 2020. Most make between a range of $54,210 and $72,902, plus employee benefits.
While numbers tend to fluctuate between cities and states, there’s no denying that these are great salaries for an entry position. Since according to one of the largest global job recruitment sites, Glassdoor, the average base salary in America is $40,000. Starting with an annual salary of $60,000 is considered uncommon and on the high end of the spectrum.
After one to two years of experience on the job, most companies provide CTAs and CRCs opportunities for professional development and promotion. Many become Clinical Research Associates, or CRAs. Indeed reports that the average salary of a CRA with one to two years of experience is $72,358. After building at least 6 years experience, a seasoned CRA should expect to earn $110,102 a year. If you would like to make more money, you can consider becoming an individual contractor CRAs. They can earn up to $300,000 in a year.
You are a science professional wanting to change careers and don’t want to go back to school
In clinical research, experience is often valued over degrees. Rather than what you didn’t study in college, hiring managers are more interested in what you have done in the past and how they can integrate you into their company.
This includes getting certified through clinical research courses, but more so what you learn from the courses you take. CCRPS offers the most in-depth CRA and CRC training so that there's tons to talk about during the interview and a working bank of knowledge during the first few months of the job. Explore more specialized certifications like ICH-GCP, Advanced Clinical Research Project Manager Certification, and Advanced Principal Investigator Physician Certification to further enhance your qualifications.
While graduate programs can help point you in the right direction, you don’t need a master’s degree to succeed in clinical research. In fact, certain positions don’t even require a bachelor’s or associate’s degree; they have certification in clinical research.
Applying to CRC and CTA positions are one of the most common segways into higher positions in clinical research. CRCs don’t need a bachelor’s or associate’s degree to get their foot in the door. While both CTA and CRA positions require a bachelor’s degree, they don’t have to be in the life sciences.
One of the best ways to gain experience and stand out from the crowd is to have on-site experience. If you need advice on how, Dan Sfera, the CEO of DSCS CRO Clinical Research Services, recommends getting started by interning or volunteering at clinics and research sites to build connections and experience.
Sometimes, the easiest way to get involved is to offer services like patient recruitment and social media management in exchange for opportunities to build your CV. By appealing to a site’s needs, this will help you get your foot in the door and build the connections and resume you need.
Another great way of adding experience to your resume is by training throughcertification courses. When employers see that you have taken the time and effort to understand how to be the best in their field, they are more far likely to hire you. At CCRPS.org, we offer seven courses and certification trainings to give you an advantage. Most of our students are hired within the first month of taking the course. We are accredited by the Accreditation Council For Clinical Research Education (ACCRE) and Joint Commission by the AMA, ANCC, and various other organizations to provide 17.5 CME credits through our CRA Certification and CRC Certification.
6. You are switching careers
Switching career fields can be nerve wracking. However, it is also an opportunity for you to be a unique candidate. Whether you come from a closely-related background, like medicine or nursing, or something completely different, there are ways you can advocate for yourself in front of employers.
If you already have a background in medicine (nonclinical doctor, unmatched MD), your knowledge of healthcare and your passion for patient health will make for a smooth translation into clinical research. In addition, your RN or MD degrees will help you gain a competitive edge and allow you to climb higher positions, such as the PI, who is the primary researcher of an operation.
On the other hand, if you come from a less relevant field but feel passionate, you can still leverage yourself to be exactly what the clinical research field needs. For example, if you are a teacher, your communication and interpersonal skills will be your keys to success. If you are a lawyer, your ability to draft and read papers will far surpass the average candidate.
If you studied mathematics, you are a skilled problem solver. If you are a translator, your language skills are valuable and will help you get into roles that require it. In short, whatever skills helped you succeed in your previous positions, you can bring it with you to clinical research.
7. You want to make a difference in disease outcomes and patient care
There are two types of people in the world: ones who accept the world as it is, and ones who strive to change it. In the last 50 years, science and medicine have gone through a series of drastic changes. However, anyone who works behind the scenes will tell you that medical breakthroughs are not miracles. Clinical research is the culmination of human effort and intelligence.
The fruits and labor of the ever-expanding industry are proof that if enough people care about the world, then they can change it. While there are many good reasons to work in clinical research, if you want the privilege to enrich the lives of others, there is a place for you in this field.
If you want to take a sneak peak at employers and opportunities near you, jobs sites like Indeed are a great resource.
Here are links for aspiring CRAs, for CRCs, and for CTAs. (Note: CTAs are often referred to Clinical Research Assistants, not to be confused for Clinical Research Associates)

How to Be a Clinical Research Coordinator
Becoming a Clinical Research Coordinator Made Easy
A clinical research coordinator, often referred to as a clinical trial manager, plays a crucial role in all kinds of medical studies. The work of a clinical research coordinator is primarily under the primary investigator, who is inherently in charge of coordinating, designing, and facilitating the clinical trial in all aspects. If you are looking to step into this role, consider enrolling in a Clinical Research Coordinator course to gain the necessary skills and knowledge.
The job of a clinical research coordinator includes:
Ensuring all processes in the clinical trial are administered smoothly.
Building relationships with sponsors, departments, and institutions to meet the demands of the investigator.
Coordinating day-to-day activities under clinical trials to ensure smooth operations.
The Main Task of a Clinical Research Coordinator
To become a clinical research coordinator, understanding the roles and duties of the position is crucial. Key responsibilities include:
Planning and managing each experiment step to keep the administrative portion of the trial under control.
Enrolling and training initiatives under clinical research while adhering to relevant laws and regulations.
Designing experiments, managing research, and facilitating medical studies across all fields.
Enrolling and screening trial participants, implementing recruitment strategies, and maintaining relationships with all participants.
Securing approval from regulatory bodies and working within both laboratory and hospital environments.
For those interested in enhancing their understanding of good clinical practices, the ICH-GCP course is highly recommended.
Qualifications You Need
Becoming a clinical research coordinator requires:
A bachelor's degree in fields like microbiology, public health administration, or medical technology.
Preferably a master's degree, especially in management studies, due to the administrative and managerial nature of the role.
Completing specific courses, such as the Advanced Clinical Research Project Manager Certification, to enhance capabilities.
Popular courses for aspiring coordinators include biostatistics, biochemistry, epidemiology, mathematics, statistics, human anatomy, and healthcare management.
Skills You Need
The role of a clinical research coordinator is dynamic and requires various skills, including:
Management and communication skills, obtainable through specialized courses like Pharmacovigilance Certification and Medical Monitor Certification.
Interpersonal skills and enthusiasm for laboratory work and multitasking.
Another crucial step in building a career as a clinical research coordinator is gaining experience as an intern in healthcare or laboratory settings.
Becoming a clinical research coordinator is a great career choice for those motivated by a combination of medicine, management, and communication. Get started now for a bright future.

7 Steps To Becoming A Clinical Research Coordinator

7 Steps to Launching Your Career as a Clinical Research Coordinator
The prospect of a career in clinical research can be exciting, especially for those with a passion for science, medicine, and helping others. A Clinical Research Coordinator (CRC) plays a vital role in this field, ensuring research is conducted ethically and efficiently. If this sounds like the path for you, here are 7 essential steps to becoming a successful CRC:
Earn a Relevant Degree:
A bachelor's degree in a science-related field like biology, chemistry, or healthcare administration is typically required (National Institutes of Health .gov). Some employers may prefer a master's degree for more specialized roles (National Institutes of Health.gov). Consider exploring the Clinical Research Coordinator course for targeted training in this role.
Gain Hands-on Experience:
Internships or entry-level positions in clinical research settings offer invaluable experience (Association of Clinical Research Professionals (ACRP)). This practical exposure strengthens your resume and provides real-world knowledge for future CRC roles. Gain further insights through the Clinical Trials Assistant Training course.
Consider Certification:
While not always mandatory, CRC certification enhances your credentials and marketability. Programs like those offered by the ACRP validate your expertise and set you apart from other candidates. Expand your certification options with the CRA course and the ICH-GCP course.
Develop Core Skills:
Crucial skills for CRCs include: attention to detail, organization, critical thinking, and effective communication. A strong understanding of research regulations and ethics is also crucial. Enhance these skills through the Advanced Clinical Research Project Manager Certification.
Build Your Network:
Attend industry events, conferences, and seminars to connect with professionals. Networking opens doors to opportunities, mentorship, and valuable industry insights. Consider further specialization with the Advanced Principal Investigator Physician Certification.
Apply for CRC Positions:
With your qualifications, certifications, and experience in place, actively seek CRC positions. Tailor your resume to highlight relevant skills and experiences, and craft a compelling cover letter showcasing your passion for research. Research the organization and demonstrate your knowledge during interviews.
Embrace Continuous Learning:
The field of clinical research is constantly evolving. Stay informed about industry trends, participate in continuing education, and pursue professional development opportunities to stay ahead in your CRC career. The Pharmacovigilance Certification and the Medical Monitor Certification can be instrumental in your continuous learning journey.
References:
National Institutes of Health (.gov): https://toolkit.ncats.nih.gov/glossary/clinical-research-coordinator/
Association of Clinical Research Professionals (ACRP): https://acrpnet.org/
Society for Clinical Research Associates (SCRA): https://www.scra.org/
A description of Clinical Research Coordinator jobs and what they entail
Clinical research coordinators are usually supervised by clinical research managers. Their main task is to administer the clinical trials. Primary responsibilities normally include administering questionnaires, informing the participants about the objectives of the study, collecting data, and managing all the trials. They also have to adhere to all trial standards that have been set and also participate in recruitment of the subjects. Clinical research coordinators also have to engage with the subjects so that they can explain the things that are expected during the trial and also find out if they have any concerns. This means that a clinical research coordinator needs communicative and interpersonal skills.
For those interested in becoming clinical research coordinators, or enhancing their skills in this role, the Clinical Research Coordinator course provides comprehensive training and certification.
The responsibilities:
Maintaining records of all studies as per the guidelines.
Sticking to all ethical standards.
Sticking to all the regulatory standards set, including those covered in the ICH-GCP course.
Administering questionnaires.
Managing the budget dedicated to the research.
Overseeing the running of the trials as smoothly as possible.
Understanding and engaging with the subjects so as to know all issues.
Making sure that all equipment and supplies that are necessary for the success of the study are working and in stock.
Participating in the recruitment efforts of the participants, a topic extensively covered in the Clinical Trials Assistant Training.
Working with the laboratories so as to share findings.
Requirements:
The qualifications of a clinical research coordinator usually depend on your locations or employer. In most cases, for you to access clinical research coordinator jobs you should:
Have an associate nursing degree or any related field
Experience of two years within the healthcare industry
Analytical mindset
Be attentive to detail
Have interpersonal skills which are exceptional
Be ready to continue learning even without being prompted to do so, which can be further supported by the Advanced Clinical Research Project Manager Certification.
Great skills in organizing
Have great verbal and written communication skills
Additional certifications such as the Pharmacovigilance Certification, CRA, Advanced Principal Investigator Physician Certification, and Medical Monitor Certification are also beneficial for those looking to further their careers in clinical research.
Understanding what clinical research organizations are and what they do
CRO or a clinical research organization is a company which operates within the pharmaceutical industry in many cases. These kinds of organizations can be involved in many processes that are involved in the development of pharmaceuticals. There are also others who only have to administer required tests on drugs that are in development.
The large companies have their own clinical research organization incorporated into the structure of the company. There are yet others who outsource drug development and testing to the organizations which are precisely designed for such a purpose.
Why hire an independent organization
When you hire a clinical research organization which is independent to handle testing, the results are not highly doubted or questioned. This is because such an organization does not have any kind of self-interest in promoting something that is clearly bad. There have been cases where some medications tested by those who make them have failed to fulfill all promises made. However, clinical research organizations have shown more value to pharmaceutical companies.
Clinical research organizations are able to pave the way for chemicals that have been successful for approval by the relevant bodies. Most bodies have very high requirements and before approval, a lot of positive data have to be provided about the chemical. Clinical research organizations can assist with the supporting documents and the necessary paperwork so that approval can go through. Individuals interested in participating in this vital role might consider enrolling in a Clinical Research Coordinator course.
There are some concerns about when and where new drugs and chemicals are outsourced to the clinical research organization, but in most cases the concerns are of an economic standpoint. Sometimes the outsourcing of chemical research organizations that are not within your country may mean that scientists within the country will have lesser jobs.
Outsourcing these services saves a lot of money for pharmaceutical and chemical companies. When they do this, the company does not need to have a clinical department maintained within. This means less strain in HR departments. For those looking to deepen their expertise in the field, courses like Pharmacovigilance Certification, CRA, ICH-GCP, Clinical Trials Assistant Training, Advanced Clinical Research Project Manager Certification, and Advanced Principal Investigator Physician Certification are available.

Clinical Research Coordinator Certification
How To Get Clinical Coordinator Certification?
The clinical coordinator or CRC is also known as the clinical trial manager. They play an essential role in the studies of medicine of all types. They work typically under the personal investigator who has a charge of managing and conducting the clinical trial at a higher level. CRC's job is to facilitate, support, and to organize the daily trial activities of the clinical. Getting a clinical coordinator certification online will take a lengthy procedure. There are some kind of the CRC at very different phases of various educational achievements. Let's look at the steps which you will need to get the clinical coordinator certification.
Step 1: Must Be Graduate From High School (4 Years)
For preparing for the clinical coordinator certification, you must begin with high school courses like biology, chemistry, and physics, also with the maths and communications. This will found your form of the foundation which will be pursued for the college studies, which also be the four-year studies.
Step 2: - Obtain A Bachelor Degree Of 4 Years
When perusing the universities and the colleges, you must focus on the courses which offer you a bachelor's degree in health sciences in terms of clinical research administration. The students should dedicate them all the things to obtain this degree and must concentrate only on this for pursuing the degree full time. These programs generally provide the clinical research professional with the tools which they will need to do the developing their medical sciences and conducting their trials and also studies. Both on the campus and the online programs on the clinical administrations focus on clinical research for placing to protect the human subjects.
The bachelor's degree can give you an entry-level position in the clinical organization or any institution. It will also help you with the existing clinicians with the advance, which is in the current jobs. The students who wish to pursue more opportunities in terms of responsibility and also the salary might also be interested in specialized training such as the Clinical Trials Assistant Training or the Advanced Clinical Research Project Manager Certification.
Step 3: - Gain Some Professional Experience
In this step, you will need to gain some experience in the field. You will have to work in clinical research with full-time research. This is the requirements for qualifying the national clinical coordinator certification. For those looking to expand their knowledge in specific areas, consider the Advanced Principal Investigator Physician Certification or Medical Monitor Certification.
Step 4: -Obtain Online
In this step, the students can get an online graduate certificate in the clinical researcher, which can be of 1 year. This will help the students to grow some knowledge around the medical world. It will help the students for getting the clinical coordinator certification. Here you can explore many types of concepts of science and designing research, data management, professionalism, and leadership. This degree will offer you strong knowledge in clinical examination, ethical research and will also provide you the experience of the medical management skill related to drugs and other trials of therapeutic. For those interested in further certification, the ICH-GCP course is also available.
Step 5: - Online Master Of Science In Clinical Research Management
This online Master of Science course is done for increasing the salary purpose of the clinical research. This is a two years course. This course includes the clinical study of coordinators and as well as the data manager of the clinical, research assistants of the social science, and also about the clinical tech laboratories. Here you can explore many types of concepts of science and designing research, data management, professionalism, and leadership. After this course, the students will get some handsome salary and also be eligible for higher-grade jobs in clinical research. This will benefit you is pursuing the clinical coordinator certification. Those interested in Pharmacovigilance may consider the Pharmacovigilance Certification to enhance their expertise in this critical area.
Get The Certification
Now it's the time to hold the accreditation of your hard work, and it determines your success. There are some categories of certifications that will help you to understand which category you fall in.

Global Pharmacovigilance Regulations
Medicine is one of the most universal forms of healthcare. However, according to the American Society of Pharmacovigilance, adverse drug events alone are responsible for $13 billion in annual American healthcare costs. The U.S Department of Health and Human Services defines adverse drug events as undesirable consequences of taking certain medications, such as “medical errors, adverse drug reactions, allergic reactions, and overdoses”. It is internationally important that a medication is professionally assessed and monitored before it is deemed safe for consumers. This is where pharmacovigilance comes into play.
Pharmacovigilance (PV), or drug safety, is the study of a drug’s adverse effects. PV helps determine if a drug is safe for mass consumption before it is put on the market. In addition, PV ensures that if drugs with serious adverse drug reactions are pulled from the market. The field is an integral part in clinical research. In this article, we will explore the various agencies and regulatory bodies that supervise PV as well as clinical research. For those interested in becoming a pharmacovigilance expert, Pharmacovigilance Certification is available to guide you through the essentials of drug safety.
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH): Created in 1990, the ICH connects global regulatory bodies and pharmaceutical companies to share technical innovations and discuss guidelines. The ICH GCP, or Good Clinical Practice, is one of the global governing clinical research guidelines for the EU, America, and Japan. Understanding and staying up to date with the ICH GCP is one of the most important qualifications someone could have before entering the clinical research industry. If you have any interest in clinical research and PV, CCRPS offers a free ICH GCP course that thoroughly explains and clarifies the complex document.
The European Medicines Agency (EMA): In the European Union (EU), the EMA coordinates PV and drug safety throughout all clinical trial phases. They ensure that a drug’s effects are monitored even after they are on the market. In addition to ICH GCP, the EU uses Good Pharmacovigilance Practice (GPvP) to determine monitoring standards of drug sales.
The Food and Drug Administration (FDA): In the United States (U.S.), the FDA supervises the approval of pharmaceutical products. Specifically, the Center for Drug Evaluation and Research or CDER is responsible for handling PV. Within CDER, the Office of Surveillance and Epidemiology assembles medical officers and safety evaluators to oversee PV in their field of experience. Most officers and evaluators are medical doctors or pharmacists.
Marketed Health Products Directorate (MHPD): The Canadian Directorate assesses and regulates health product risks. They are composed of 6 different bureaus and offices, each with their own area of speciality. Together, the Directorate monitors adverse drug effects and makes regulatory decisions.
Pharmaceuticals and Medical Devices Agency (PMDA): Established in 2004, the Japanese regulatory authority PMDA supervises the safety of drugs from the lab to the market. Not only do they consult clinical trial professionals on clinical compliance, they also provide post-market safety measures and relief services for adverse health effects.
Since drug safety is important for patient health and safety around the world, there are career opportunities for PV virtually anywhere. If you want to learn more about pharmacovigilance, please visit our website at CCRPS.Org. Our online Pharmacovigilance Certification guides new professionals to improve their qualifications and gain valuable insight. The course is curated by real clinical research professionals and flexible enough to fit any schedule. Moreover, for those looking to expand their career into clinical research coordination or clinical trials assistance, CCRPS offers comprehensive courses such as the Clinical Research Coordinator, Clinical Trials Assistant Training, CRA certification, Advanced Clinical Research Project Manager Certification, Advanced Principal Investigator Physician Certification, and Medical Monitor Certification.

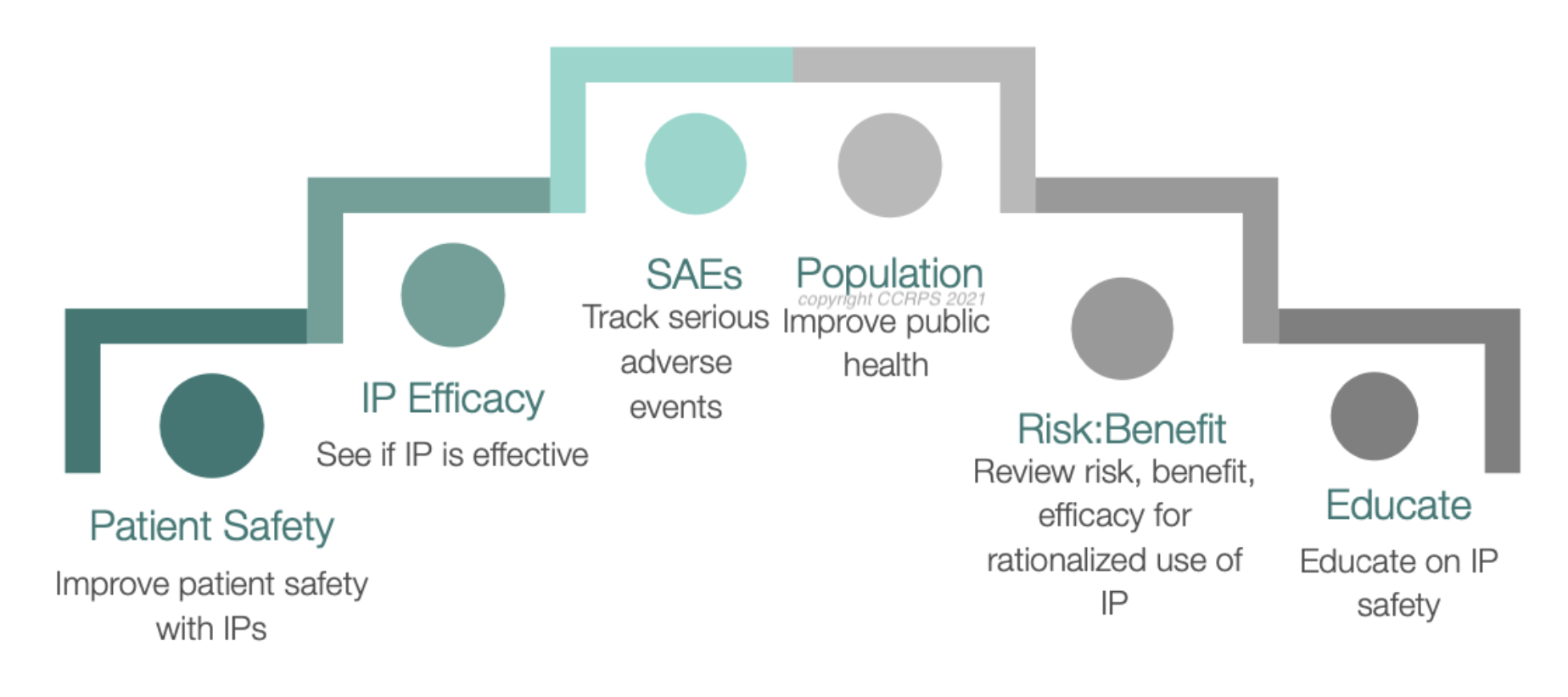






What Is a Clinical Research Associate?
Clinical Research Associates are those responsible for the planning, setting up, and coordinating of clinical trials. They provide technical assistance for experiments, make sure that the scientists that are involved in the clinical research trials comply with regulatory standards, and also collect results during the clinical trials. When the trials are over they are also to be involved in the presentation of the results to the public in a manner that is useful and understandable.
Clinical research associates are those responsible for the planning, setting up, and coordinating of clinical trials. They provide technical assistance for experiments, make sure that the scientists involved in the clinical research trials comply with regulatory standards such as ICH-GCP, and also collect results during the clinical trials. When the trials are over, they are also involved in the public results presentation.
The amazing part about being a clinical research associate is that jobs are available for them in both the laboratory and office settings.
SPECIALIZATIONS
They focus on the following:
Designing and implementing clinical research trials
Training staff, which could be enhanced by taking the Clinical Trials Assistant Training course.
Monitoring progress
Screening test subjects
Presenting findings
Helping maintain the database of all clinical research trainees
SKILLS
As a clinical research associate, you should have the following skills:
Communication skills
Science knowledge
Strong mathematics knowledge base
These core skills are what you need in the clinical research profession.
EDUCATIONAL REQUIREMENTS
As a clinical research associate, you should have one of the following:
High school diploma and 6,000 hours of experience.
An associate degree in clinical research fields and 4,500 hours of experience.
A Bachelor's, Master's, or Registered Nurse degree and 3,000 hours of clinical research experience.
You can come from a variety of medical sciences or health-related fields, or from a nursing background as an RN. Courses offered in hospital and clinical related ethics, team management, and research methodologies will be valuable to your education. Consider advancing your career with certifications like the Advanced Clinical Research Project Manager Certification or the Advanced Principal Investigator Physician Certification.
SALARY
The median salary of a clinical research associate boasts of $90,310 annually and it is always on the rise. Plus, there is always a ready market for clinical research professionals.
Explore more about this field with specific courses like the Clinical Research Coordinator training, Pharmacovigilance Certification, or the CRA course. These certifications will equip you with the necessary skills and knowledge to excel in this growing industry.
Take courses from CCRPS and learn more on how to become a clinical research professional.
Discover more from Clinical Research Training | Certified Clinical Research Professionals Course

CITI Clinical Research Coordinator Course

Significance of CITI Clinical Research Coordinator Course
The diseases that impact the world need solutions. To find aid for the physical and mental issues of this generation, the group of experts in the medical field work day and night. However, they can’t do this on their own. They need clinical research coordinators to help them find success. In this article, you will get to know the significance and responsibilities of clinical research coordinators, and why programs like the CITI Clinical Research Coordinator Course are critical to aspiring professionals and the field as a whole.
Who is a clinical research coordinator?
Basically, a clinical research coordinator works with a principal investigator in medical research. The principal investigator is responsible for large aspects of the project, such as securing grants and writing protocols, while the clinical research coordinator is responsible for the day-to-day operations of research. Some tasks include:
Maintaining records and documents
Recruiting patients
Ensuring trials are following protocol
Keeping the principal investigator informed on developments
Managing supply inventory
The clinical research coordinator needs to complete a variety of tasks, but overall they need to have strong communication and organization skills. For those looking to advance in this role, the Advanced Clinical Research Project Manager Certification might be of interest.
More about the Clinical Research Coordinator Course
Being a clinical research coordinator is not easy. Thus, education and re-education is critical to field professionals.
Many different institutes and universities all around the world offer courses, but the CITI clinical research coordinator course stands out. The course is balanced and well-thought out. Students get to learn about ethics as well as the responsibilities of the clinical coordinator. The institutes do not only teach textbook knowledge, but they also encourage practical knowledge. For a comprehensive learning experience, students might consider exploring related certifications like Pharmacovigilance Certification or Clinical Trials Assistant Training to gain insights into different aspects of clinical trials. Those in more specialized roles may find the Advanced Principal Investigator Physician Certification and Medical Monitor Certification to be valuable additions to their educational pathway.
If you feel inspired, check out their course page here. If you want to compare options, try CCRPS. We offer affordable, ACCRE accredited courses dedicated to clinical research coordinators. Similarly, our courses are written by real professionals that know how to help someone stand out in the field. Still not sure yet? Below are some of our articles that will help you better understand clinical research and make an informed decision.
Take courses from CCRPS and learn more on how to become a clinical research professional.
Discover more from Clinical Research Training | Certified Clinical Research Professionals Course

Clinical Study Coordinator Training Program
A study coordinator is the backbone of any research project. They are responsible for ensuring that all the elements of the study further the goals of the study, or the research objective. A study coordinator’s foresight and attention to detail are critical to the success of every trial.
Role Of A Study Coordinator
The role of a study coordinator is broad and includes the following:
In the planning stage: ensure that the plan is comprehensive enough to cover all aspects of the study and that various processes and elements of the study are linked. For those looking to become a study coordinator, the Clinical Research Coordinator course can provide essential training.
At the time of execution: ensure the availability of funds, apparatus, infrastructure, etc. required for the study. Further, they also have to ensure that the study flows promptly. Knowledge of ICH-GCP standards, covered in our certification course, is crucial here.
When various elements of the study are near to completion: the study coordinator will have the role to align them in a planned manner.
At all stages for research: a study coordinator ensures that research ethics are maintained, and there is no violation of any code of conduct.
Determine the Standard Operating Procedures (SOP) to be followed in a study. The Advanced Clinical Research Project Manager Certification can help develop the skills needed to define and manage SOPs effectively.
Work opportunities for a study coordinator
In many countries, such as India, study coordinators are a relatively new career route and hence acutely unexplored. However, considering the growth in research and development areas, it is pretty evident that there is an increasing demand for them. Presently, the industry needs study coordinators while only a handful of them exist.
Due to this demand-supply gap, the monetary compensation of such positions has been very high. Research by Salary.com contends that the median salary of a clinical research coordinator is around $63,330. However, based on the type of study, the range may vary from as low as $2,000 per month to as high as $4,500 per month. Those interested in expanding their career options might consider training as a Clinical Trials Assistant, or specialize further with the Advanced Principal Investigator Physician Certification or Medical Monitor Certification.
Importance
Every field of work needs consistent education, and there's always a scope for enhancement. The area of work of a study coordinator is no exception. The sky is the limit, and each training program is a step towards reaching perfection.
A study coordinator training program focuses on developing individuals to perform the roles mentioned above, in addition to other roles that may be expected from a study coordinator.
For example, regulations and code of ethics governing the respective field of study may also get updated from time to time. In such cases, it becomes necessary for the coordinators to obtain a basic understanding of the changes and implement them in a study.
Moreover, certain organizations may require applicants to have taken certain training courses before considering them for a position. Thus, it has become evermore important for hopeful professionals to take the right training courses.
Who Will Provide The Training?
Depending on the area of study, various institutions, or people with immense experience in the relevant field may provide training.
For example, an organization may organise EA training for its employees from experts in the industry. Other examples of are specialized training courses for coordinators, such as the Clinical Research Coordinator (CRC) training provided by the CITI Program.
Further, various universities run comprehensive and dedicated courses for imparting study coordinator training. Some such institutes are Clinical and Translational Science Institute (CTSI), which provides basic coordinator training, and ACRP, which provides Certified Clinical Research Coordinator (CCRC) training.
Conclusion
The field of a study coordinator is a career that has immense potential yet to be explored. More trained professionals can prove to be very valuable assets to the research processes. At CCRPS, we offer ACCRE accredited training specialized for study coordinators. In addition, we have complied articles below to help you better understand the complex aspects of clinical research.
Take courses from CCRPS and learn more on how to become a clinical research professional.
Discover more from Clinical Research Training | Certified Clinical Research Professionals Course

How to Save Money on Becoming a Clinical Research Coordinator (CRC)

If you think practically, then you will find that nothing in this world comes for free. But if you have a passion for clinical research and need to learn on a budget, here are some ways you can enhance your understanding without breaking the bank.
To understand how you can learn for free, it’s first important to understand the typical trajectory of a clinical research coordinator:
A person seeking clinical research coordinator training should complete high school. There they learn must-have all the science subjects like physics, chemistry, and biology.
After high school, institutes for professional clinical research coordinator professionalism offer programs that are essential for later.
In addition, one can get an experience graduate certificate from an online source. This would help the person reach their career goals faster.
Alternatively, they can complete a bachelor's degree of science.
After completing the bachelor's degree, a master's degree is needed for some of the higher pay-grade positions.
How to get free clinical research coordinator training?
If looking at the education requirements for a clinical research coordinator makes you dizzy, you’re not alone. Becoming a clinical research coordinator takes a lot of time and money. That is why many students turn to scholarships and healthcare programs to help pay their tuition.
On the other hand, there are many websites that offer free or affordable information and training for aspiring professionals. While they can’t replace a formal education, they can supplement your resume and knowledge. While some important topics include clinical data management, pharmacovigilance, and regulatory authorities, you should strive for a comprehensive understanding of the field. At CCRPS, we offer affordable courses designed for clinical research coordinators as well as free ICH GCP training. These can help you build your resume and land the position you want.
In addition, it is really important to keep updated with new clinical research headlines. Below, I have complied some articles that aspiring professionals might find useful. The best thing you can do for your education is to start now and not later.
Take courses from CCRPS and learn more on how to become a clinical research professional.
Discover more from Clinical Research Training | Certified Clinical Research Professionals Course




How to Make a Career out of Clinical Research Management (CRC)?
A clinical research coordinator or CRC is a highly trained professional, whose expertise is crucial to all medical research work. Although a CRC works under the constant guidance of a principal investigator, they are responsible for the day to day trials and clinical operations. Thus, this profession can be quite challenging but rewarding.
However, getting into the crew of CRCs is not an easy task. A particular individual may need to prepare for a while to be selected for such a post. In this segment, you can learn all the aspects to becoming a successful CRC.
What does a Clinical Research Coordinator do?
A clinical research coordinator or CRC is a highly trained professional, whose expertise is crucial to all medical research work. Although a CRC works under the constant guidance of a principal investigator, they are responsible for the day-to-day trials and clinical operations. Thus, this profession can be quite challenging but rewarding.
However, getting into the crew of CRCs is not an easy task. A particular individual may need to prepare for a while to be selected for such a post. In this segment, you can learn all the aspects to becoming a successful CRC through specialized training such as the Clinical Research Coordinator course.
What does a Clinical Research Coordinator do?
The field of medicine is always advancing. In the world of medicine, new knowledge is indispensable. So, the professionals of the medical branch are always looking to conducting experiments that can lead them to new cures and successes. Among clinical research professionals, clinical research coordinators are at the front lines of research operations and analytics.
The duties of a CRC are to plan and initiate a study or experiment the board has approved. During the phases of the project, they must maintain communication between their site and institutional organizations. The CRCs are eligible to do the research and reviews. Additionally, they can choose to hire potent candidates for their purpose. Understanding and adherence to ICH-GCP guidelines are crucial for ensuring compliance and ethical conduct in their studies.
How can you be a successful clinical research coordinator?
The job of a clinical research coordinator is not an easy one. However, with enough hard work and patience, one can become eligible for the post. Although, individuals who aspire to become a clinical research coordinator are advised to understand the basics and keep educating themselves if they want to become a trusted professional. Continual education and certification, such as the Pharmacovigilance Certification and Clinical Trials Assistant Training, can provide essential knowledge and skills required in this evolving field.
Additionally, for those looking to further their careers, the Advanced Clinical Research Project Manager Certification and Advanced Principal Investigator Physician Certification are excellent resources to consider. Those interested in overseeing clinical trials and research safety might consider the Medical Monitor Certification.
Here are the requirements to become a successful clinical research coordinator:
1. To apply for this post and profession, the candidates need to have a background that is related to the field of medicine or biology. The general preference for applying is to have a bachelor's degree in microbiology or medical technology.
2. Some positions require individuals who have a master’s degree on top of their bachelor’s degree. So, you might need to invest more time into your education if you want to work with certain firms or reach a higher pay-grade.
3. Preparing yourself for the job is crucial. As this job is a challenging one, individuals might need to develop and train themselves for quite some time to do well. For young training candidates, clinical trial coordinator training is shown to be quite a useful resource. At CCRPS, we offer affordable training courses developed by real professionals and accredited by ACCRE. If you are looking to learn more about clinical research and specific positions, CCRPS can be the perfect starting point.
A step towards the development of humankind
Medical science has experienced great expeditions over the past few decades, and all of that has help benefit society in some way or the other. Thus, becoming a part of a research team is a commendable task indeed.
Becoming a clinical research coordinator isn’t easy. A CRC must have an eye for detail and need to continually polish their skills. Moreover, experience and education is a determining quality that can set a good CRC apart from the rest in the lot. If you’d like to start your education, check out CCRPS. Our courses and articles will help you find the tools to succeed in clinical research.
Take courses from CCRPS and learn more on how to become a clinical research professional.
Discover more from Clinical Research Training | Certified Clinical Research Professionals Course
Clinical Research Coordinator Classes
A Guide to Clinical Research Coordinator Classes
The medical field thrives on constant innovation. From uncovering new treatments to battling life-threatening illnesses, there's a relentless need for fresh talent. One crucial role at the forefront of medical discovery is the clinical research coordinator (CRC).
Who is a Clinical Research Coordinator?
A CRC, sometimes called a site research coordinator, study coordinator, or simply CRC, is the backbone of a research site. You'll possess a deep understanding of research guidelines, clinical processes, and more. Your responsibilities include documentation, subject well-being, and conducting research procedures. To develop a foundational understanding, consider enrolling in a Clinical Research Coordinator course.
What Does a Clinical Research Coordinator Do?
A CRC's role requires meticulous attention to detail and unwavering precision. CRCs are guided by a principal investigator (PI), who oversees the entire research project's proper execution.
The studies CRCs undertake are often complex, requiring days or even months of analysis. Patience and focus are essential qualities. Moreover, as a representative of your medical institution, PI, and colleagues, building strong relationships with other professionals is paramount.
Building a Fulfilling Career as a CRC
There's no single educational path to becoming a CRC. A background in pharmacy, nursing, business administration, statistics, biology, teaching, health record maintenance, or even medical technology can pave the way. CRCs find employment in research groups, private institutions, pharmaceutical companies, biotechnology firms, and more.
The Skills and Knowledge of a Successful CRC
Clinical research demands accuracy. Refining your skills is crucial for success. Here's what you can leverage when applying for CRC positions:
Educational Background: A bachelor's degree in microbiology or medical technology is ideal. However, relevant experience and coursework can strengthen your application. Employers value what you bring to the table, not what you lack.
Experience: Entry-level positions often seek candidates with 1-2 years of experience, while senior roles might require 5-6 years. Master's degrees can be advantageous for higher-level positions with better pay.
Enhancing Your Qualifications: Clinical Research Coordinator Classes
Formal education through clinical research coordinator classes can significantly enhance your qualifications. These programs equip you with the specific knowledge and practical skills required to excel in this dynamic field. Here are the different types of CRC classes available:
Certificate Programs: These intensive programs offer a comprehensive foundation in clinical research principles, regulations, and best practices. They typically last several months and can be completed online or in-person. For those interested in gaining specialized knowledge, exploring a Pharmacovigilance Certification or ICH-GCP course can be highly beneficial.
Associate's Degree Programs: For those seeking a more in-depth education, associate's degree programs delve deeper into research methodology, data management, and ethical considerations. They can take up to two years to complete. Aspiring research coordinators may also consider a Clinical Trials Assistant Training program to further enhance their practical skills.
Bachelor's Degree Programs: A bachelor's degree in clinical research provides the most thorough education. These programs equip you with advanced research skills, project management expertise, and a strong understanding of research ethics. Earning a bachelor's degree can take four years or more.
Benefits of Taking Clinical Research Coordinator Classes
Investing in CRC classes offers several advantages:
Stronger Job Prospects: Formal education demonstrates your commitment to the field and equips you with the knowledge and skills employers seek.
Enhanced Skills and Knowledge: You'll gain a comprehensive understanding of research protocols, data collection, regulatory compliance, and ethical considerations.
Career Advancement: Formal education can open doors to senior-level positions and better career opportunities.
Networking Opportunities: Many programs offer opportunities to connect with instructors and fellow students, building a valuable professional network.
For those aiming at leadership roles or seeking to further specialize, consider advanced certifications such as the Advanced Clinical Research Project Manager Certification or the Advanced Principal Investigator Physician Certification. Additionally, a Medical Monitor Certification can prepare you for critical oversight roles within clinical trials.
Finding the Right Clinical Research Coordinator Class
When choosing a CRC class, consider these factors:
Accreditation: Ensure the program is accredited by a reputable organization.
Course Curriculum: Evaluate if the curriculum aligns with your career goals and covers essential topics like research ethics, Good Clinical Practice (GCP), and regulatory requirements.
Delivery Format: Choose between online, in-person, or blended learning options to suit your learning style and schedule.
Cost and Time Commitment: Consider the program's cost and how long it will take to complete.
Conclusion
A career as a clinical research coordinator is a rewarding opportunity to contribute to medical advancements. By taking advantage of clinical research coordinator classes, you can gain the knowledge and skills to thrive in this dynamic and growing field. With dedication and the right education, you can launch a fulfilling career at the forefront of medical discovery.
Additional Tips
Research professional organizations: Explore resources offered by organizations like the Association of Clinical Research Professionals (ACRP) for career guidance and educational opportunities.
Volunteer in research settings: Gain valuable practical experience by volunteering for research studies or clinical trials.
Develop transferable skills: Hone your communication, interpersonal, and organizational skills.
Feel free to check out our courses and some of our other articles in the slider below.
Take courses from CCRPS and learn more on how to become a clinical research professional.
Discover more from Clinical Research Training | Certified Clinical Research Professionals Course